
Hello, everyone! I am working with pseudo bulk RNA-seq data and facing challenges with designing an appropriate analysis approach due to confounded batch effects and unbalanced conditions. Here is a summary of my data.
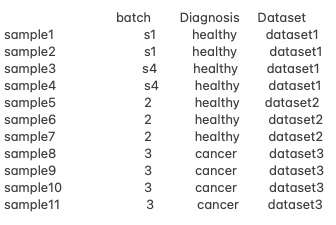
challenges:
- The Diagnosis groups (e.g. healthy vs. cancer) do not overlap with the same batches, making it impossible to adjust for batch effects using the typical design matrix:
~ Diagnosis + Batch. - I'm interested in comparing healthy vs. cancer samples while eliminating batch effects.
Questions:
Is there an alternative model or approach in tools like edgeR, limma-voom, or DESeq2 or any other that can handle confounded batch effects? (Currently I'm working with edgeR with passing Diagnosis as a single factor to the design matrix. But MDS plot separate clusters for dataset1, dataset2 and dataset3)
Would combining Diagnosis and Batch into a single group factor be advisable here?
Are there any tools that take preprocessed(batch-corrected data, i.e. I have )data in differential expression analysis? (I guess edgeR only works with raw counts)
Thank you in advance for the help.

Thank you for your reply. I currently have batch-corrected data processed with scvi-tools (specifically, totalvi) and have followed the same pipeline for analyzing reconstructed counts in edgeR as I did for raw data. However, I am observing differences in the TopTags output, particularly among the top 100 genes when ordered by p-value.
It also appears that edgeR might be designed primarily for discrete count data, which could pose challenges when using it with reconstructed data that might be more continuous in nature.
Have you read the totalVI documentation?, which says:
If you used totalVI, then you must use it for the DE analysis as well, because it does not pass useable expression values on to other programs. I have no faith however that totalVI can correct for completely confounded batch effects or that it can return realistic p-values or posterior probabilities taking into account biological variation.
Trying to analyse totalVI output in edgeR would be nonsensical. It is not a matter of discrete vs continuous (edgeR is perfectly capable of analysing continuous expected counts from RSEM, Salmon or kallisto), but rather the fact that effects like library size and donor effects have been removed and the resulting data is on the wrong scale.
Thank you for sharing your insights. I truly learned a lot from your explanations!