
Hi,
I have use SGSeq mostly to look at exon usage for genes of interest in my RNASeq datasets. I would like to move toward exon diffex analysis. I followed the recipes in the HTML guide to get counts, exons and gene ID but I am perplexed about what I got... I have an SGFeatureCounts object with created from rowRanges on the SGVariantCounts object previously created that has all the infos like geneName, etc. However, after performing the getSGVariantCounts method on it and using the eventID method, I only get integers, that I am guessing are the positions of the genes in the previous object. How can I get geneName instead or possibly add geneName data on the objects?
I am guessing that it would always be possible to go back with integer values to explore the SGFeatureCounts object?
Thanks in advance
S

Hi Leonard,
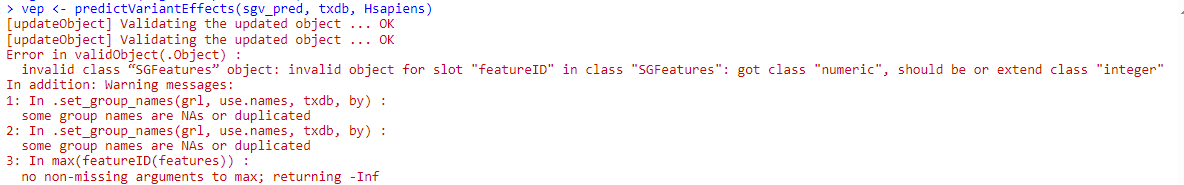
No problem ;-) Ok, as per section 11, I created objects for counts, exon ID and gene ID with the methods in the vignette:
# My R stuff: # sgfc.denovo.var is my equivalent to sgvc_pred > sgv.ranges<-rowRanges(sgfc.denovo.var) > sgv.counts<-getSGVariantCounts(sgv.ranges,sample_info=data.r) > x <-counts(sgv.counts) > vid<-variantID(sgv.counts> > eid<-eventID(sgv.counts)When I look at vid or eid, I only get matrices of integer values, so I am guessing that they are corresponding indexes in the previous sgv.ranges object. Am I right? If this is the case, what is the easiest way to make out the gene symbol from the geneName column in sgv.ranges?
Thanks in advance
S
Hi Sylvain,
Yes variantID() and eventID() are accessor functions for metadata columns in the SGVariant/Counts objects. You can see all available columns by typing mcols(sgv.counts). To extract gene names you can simply use geneNames(sgv.counts). Note this will return an empty CharacterList unless you previously annotated your predictions. I hope this helps.
Leonard
Hi Leonard,
Thanks for the info; I'll be playing some more and if needed, may be bug you again ;-)
Best regards
S
Hi,
I was following the SGSeq tutorial, but there were some things that I didn't understand properly. I would appreciate further explanation, please.
As following the tutorial document of SGSeq for section 11, I created the SGVariantCounts object:
Then, the plan was to use DEXSeq for differential splice variant usage.
What information do I have to extract and use as input for differential splicing analysis using DEXSeq? How do I build my DEXSeqDataSet object for further analysis?
In the documentation of DEXSeq they used DEXSeqDataSetFromSE to build DEXSeqDataSet object as follow:
Now, as I used SGSeq; what do I have to use to create the DEXSeqDataSet object? Should I use the DEXSeqDataSetFromSE function, and replace see with x which is the counts(sgvc)? or there is another way? How I have to make my design? use vid from SGSeq instead of exon in the dxd from DEXSeq?
I suppose I have to use my SGVariantCounts objects in my SGSeq output as an input in DEXseq like this, but I am not sure:
but then here how to define sample and exon?! or where to get these info from my SGSeq analysis?
I would appreciate any help!
Many thanks in advance!