
Hello!
I am new to the analysis of ChIP-seq experiments, so please be gentle if I did make a stupid mistake.
I am trying to identify consensus peaks in a series of 6 ChIPs with different antibodies for the same target (H3K27ac) in the same cell line. I used bbmap to map reads to hg38, and MACS2 to find narrow peaks and generate FE bedgraphs.
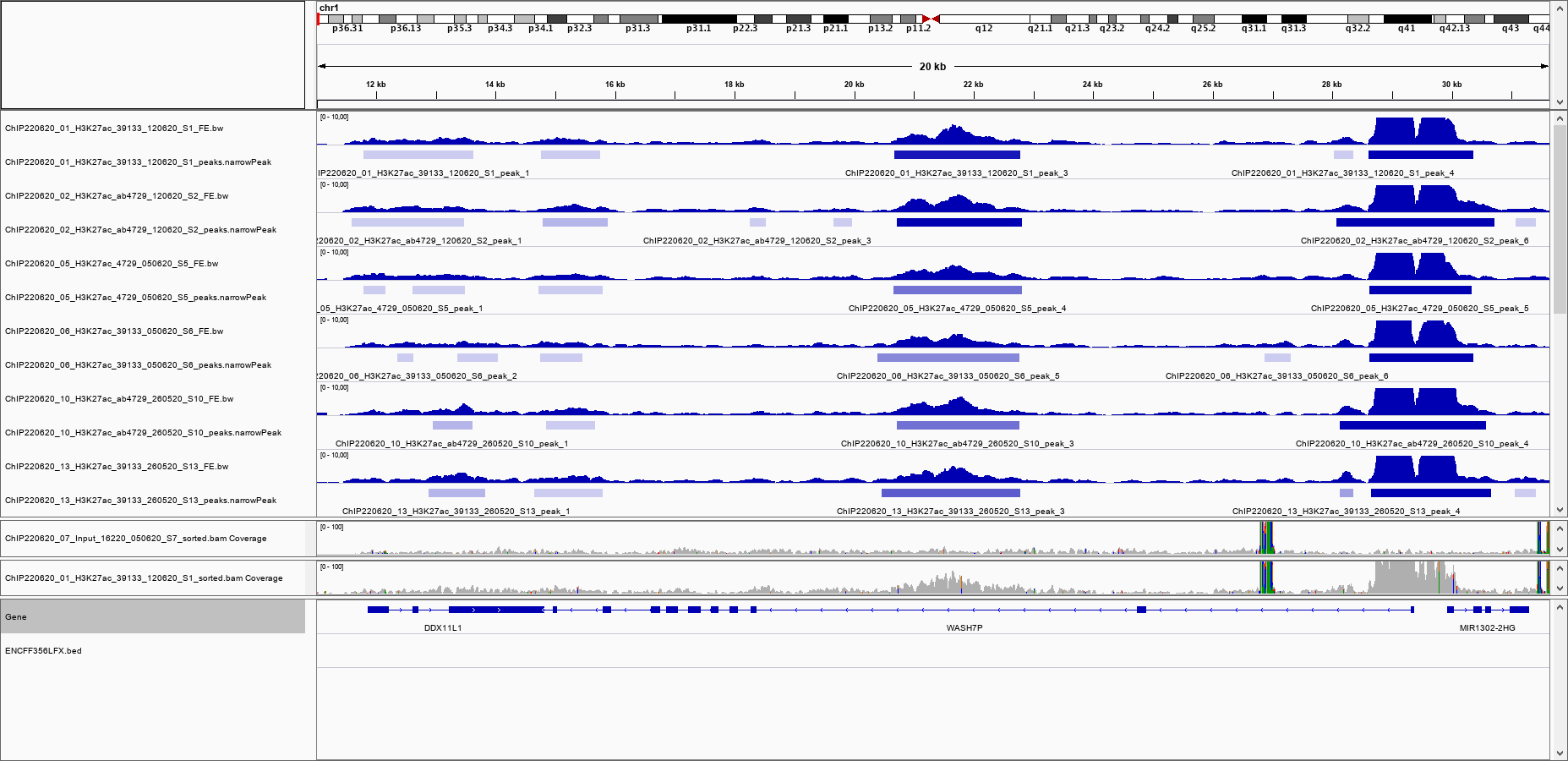
I inspected the pileups visually (IGV snapshot attached), I see 3 peaks in common to all 6 samples: a weak peak at chr1 around 15.000 bp, a medium strong peak at around 21.500 bp, and a strong peak at around 29.000 bp. All 3 peaks also show up in all 6 .narrowPeak files.
When I run the following code:
library(DiffBind)
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
samples <- read.csv("meta/samplesheet.csv")
dbH3K27ac <- dba(sampleSheet = samples) ## Sample sheet with bam files, bam control files, narrowPeak files
dbH3K27ac <- dba.blacklist(dbH3K27ac, blacklist="BSgenome.Hsapiens.UCSC.hg38", greylist=FALSE)
dbH3K27ac <- dba.count(dbH3K27ac, summits=T, minOverlap = 0.66, bUseSummarizeOverlaps = T)
dbH3K27ac_consensus <- dba.peakset(dbH3K27ac, bRetrieve = T, writeFile = "H3K27ac_consensus")
the resulting file is missing the peak at 21.500 bp:
chr1 14652 15878
chr1 28036 30721
...
I have attached the IGV snapshot to illustrate. In the IGV panel, I have also loaded the BAM of the input and of one of the ChIPs, just to exclude that there is anything in the input file in the region of question. The IGV snapshot also shows the content of the blacklist file, which shows that the region around 21.500 bp is not blacklisted.
Why is the peak at 21.500 bp gone? I am worried that there are more peaks that disappear, and that my analysis will not show the complete picture in the end.
Thank you for any input!
Best regards Sebastian
> sessionInfo()
R version 4.0.3 (2020-10-10)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 17763)
Matrix products: default
locale:
[1] LC_COLLATE=German_Germany.1252 LC_CTYPE=German_Germany.1252 LC_MONETARY=German_Germany.1252
[4] LC_NUMERIC=C LC_TIME=German_Germany.1252
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] TxDb.Hsapiens.UCSC.hg38.knownGene_3.10.0 GenomicFeatures_1.42.0
[3] AnnotationDbi_1.52.0 BiocParallel_1.24.0
[5] DiffBind_3.0.1 SummarizedExperiment_1.20.0
[7] Biobase_2.50.0 MatrixGenerics_1.2.0
[9] matrixStats_0.57.0 rtracklayer_1.50.0
[11] GenomicRanges_1.42.0 GenomeInfoDb_1.26.0
[13] IRanges_2.24.0 S4Vectors_0.28.0
[15] BiocGenerics_0.36.0 forcats_0.5.0
[17] stringr_1.4.0 dplyr_1.0.2
[19] purrr_0.3.4 readr_1.4.0
[21] tidyr_1.1.2 tibble_3.0.4
[23] ggplot2_3.3.2 tidyverse_1.3.0

Hello Rory,
thanks for your help. The dba object can be downloaded here https://drive.google.com/file/d/14ZBmX2PadnjrZpV7Fi-oBxip2uYwrVFl/view?usp=sharing.
The peak is present in all 6 samples before blacklisting:
As you suggested, I have double-checked both blacklist and the blacklisted peaks, and it's not in there:
Thank you very much for troubleshooting!
Best
Sebastian
Could you run
dba.count()withfilter=0and send me the result as well?Sure. Please find the dba object here: dbH3K27ac_filter0.rda
The peak seems to be in there now.
Thank you!
Yes, the peak is there, but it has very few reads in either the ChIP or Control, even though we see pileups in the browser screenshot:
Without the bam files I'm somewhat limited in what I can try. You can make them available (you can send the link in email instead of posting it here), or try a couple of things yourself:
dba.countwithmapQCth=0(andfilter=0), then see if the read counts change (as above).This will check if the reads are being filtered. The peak is very close to the start of the chromosome, and I'm wondering if the alignments have low quality scores. I think this region was in the blacklist once upon a time, and looking at your control track, it would likely be filtered out if you ran a greylist.dba.count()withsummits=FALSE(andfilter=0), then see if the read counts change (as above). This will check if there is an issue with the read method (summarizeOverlapsis not actually used whensummits=TRUE) -- if so we can work around that easily enough.Thank you very much for your help. I'm very sorry for the late reply, I was busy with clinical duties...
I've tried the two things you suggested:
Does this mean that the mapping quality is very low at this peak? I tried to assess mapping quality at this location in IGV, and when I click on individual reads, it shows a MAPQ of 3 for any of the reads that I've clicked (see attached image). MAPQ is also 3 for any of the reads in the peaks at 15000 and at 29000.
I'll try to cut the BAM file down and make them available for download later.
Thank you for your help!
This makes sense. MAPQ of 3 indicates a multi-mapped read -- there is not a unique alignment. This may be a repeat region which is why is has shown up in blacklists before (difficult to get consistent reads unambiguously mapping to this region).
The default behavior in
DiffBindis to ignore multi-mapped reads, however by settingmapQCth=0you are including the "primary" mapping. In my experience, unless there is some reason to be paying particular attention to problematic repeat regions, it is cleaner to filter them out. (I have have had to deal with this case a few times, and put more effort into disambiguating the reads, but it adds a lot of work!).Bottom line: unless you have some reason to be particularly interested in this near-telomeric region, you can ignore it (so long as you are getting positive read counts along most of the genome).
This absolutely makes sense. I don't really want to include these peaks in my analyses.
Thank you so much for your help!!
Best wishes
Sebastian