Entering edit mode

ahua217
▴
10
@ahua217-24323
Last seen 4.9 years ago
Hello!
I met a problem when I tried to construct DBA object with dab() in Diffbind.
This is my sampleSheet.
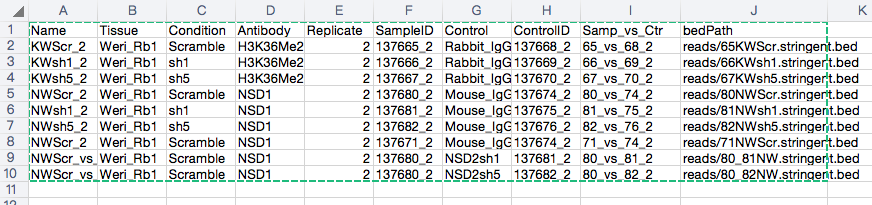 And the command
dbaNSD <- dba(sampleSheet="sampleSheet.csv", dir="/Users/ahua217/AAData/Oct17/peak/DiffbindNSD")
And the command
dbaNSD <- dba(sampleSheet="sampleSheet.csv", dir="/Users/ahua217/AAData/Oct17/peak/DiffbindNSD")
returned an error
137665_2 Weri_Rb1 Scramble 2 raw
Error in if (is.na(peaks)) { : argument is of length zero
I then used "cat -v" linux command to compare my sampleSheet to that in Diffbind vignette but did not find any format difference I can change. After exhausting some adjustments I could think, I posted the problem here for help. There seems no place to upload my sampleSheet.csv, but it is indeed a comma-delimited .csv file, generated from WPS.
Thank you very much!

Thank you so much Dr. Stark. I used to think that the DBA object could be generated with only parts of those columns and could be edited later. I will try with your suggestion tomorrow. My data were from CUT & RUN software SEACR, which gave out bed files. SEACR already provided control option of IgG and generated the bed outputs. So I thought I should not include bamControl again in Diffbind, right?
The .bed files output by
SEACR(the ones you have in thebedPathcolumn) contain the enriched regions (peaks). So they should be specified in thePeakscolumn. You should also include the original bam files (the ones used to create the bedgraphs input toSEACR) for each sample in thebamReadscolumn.You do not need to include the IgG controls in the
DiffBindsample sheet.I did it again by adding what you suggested. I am happy the sampleSheet is successfully loaded through dba() now. Thank you very much! Now I have two questions with it: 1) SEACR asked for bedgraph files as input. So I filled the bamReads column with the bedgraph files but not original bam files. Does that matter? Those bedgraph files were transformed from the bam files with scale factors through bedtools. Since the scale factors were generated from the reads of exogenous genome in CUT & RUN protocol and not 1X, I thought the bedgraph files contained more information than the bam files for determining peaks in Diffbind. Is that right? 2) Can I do something with the "peakCaller" parameter in dba()? For example, adding "SEACR" as an peakCaller? Is the default setting of peakCaller suitable in my case? Thanks!
DiffBindrequires the original bam files, it does not work on bedgraphs. Re-scaling/normalizing will be done for differential analysis withinDiffBind. It sounds like to have exogenous "spike-in" chromatin, right? If these was aligned separately, you can specify the spike-in bam files using a 'Spikein' column in your sample sheet and specifyspikein=TRUEwhen callingdba.normalize(). If they were aligned together in a combined reference genome, you can specify the names of the exoegenous chromosomes as thespikeinvalue when callingdba.normalize().If you do not specify a
PeakCallerin the samplesheet, the default peak score ('raw') will be to use the fourth column of the output, which for SEACR is the total signal for the peak. You could also set it to 'bed', which would give you the fifth column, which is the maximum signal. It really doesn;t matter which of these you use as the peak scores aren't very important as they are over-ridden by count-based scores after callingdba.count().Thank you Dr. Stark. My peaks were called using SEACR, with both target files and IgG files. Before SEACR, all of them were separately processed with their own scale factors, which were calculated with spike-in genome reads. So my samplesheet now has such columns: "peaks", "bamReads"(original target bam files), "bamControl"(original IgG bam files) and "Spikein"(scale factor corresponding to bamReads), right? Do I need to include a 2nd "Spikein" column which is corresponding to IgG bam files? Thanks!
The
Spikeincolumn in the samplesheet should only be set to bam files, never to scaling factors. You don't need to worry about the IgG control for the spikein reads. If you want to use pre-computed scaling factors you can add them usingdba.normalize()and thenormalize=(and possible thelibrary=) parameter without specifyingspikein=so thatDiffBindwill use the factors you supply rather than re-computing them.Even if you used SEACR to call peaks with pre-computed scaling factors based on the spike-in reads, you can still have
DiffBindcompute its own spike-in scaling factor to do a differential analysis of those peaks. How to do this depends on if the spike-in reads, which I assume come from an different genome, were aligned into separate bam files, or the primary and spike-in reference genomes were concatenated such that both the main reads and the spike-in reads are in aligned together into the same bam file (but along different chromosomes).If you have different bam files for the spike-ins, they should be listed in the
Spikeincolumn. If the spike-ins are included in the main bam files, you don't need aSpikeincolumn; when you calldba.normalize(), specify a the names of the chromosomes the spikein reads are aligned to as the value for thespikein=parameter.Thank you Dr. Stark! I feel much clear after practicing some functions you suggested. My remaining problem is my Diffbind could not find the function "dba.normalize". Is it due to a version issue? I just installed Diffbind last month.
Tou didn't include your output from
sessionInfo(), but I assume you are running older versions of R and Bioconductor.dba.normalize()was introduced in Bioconductor 3.12 (DiffBind_3.0.7).To use the latest version, you need to update to R version 4.0.3 and then install the current Bioconductor version using
BiocManager::install(version = "3.12").Thank you Dr. Stark for your timely help every time! Now my "dba.normalize()" function is active.