
Hello! I have a DBA object like below
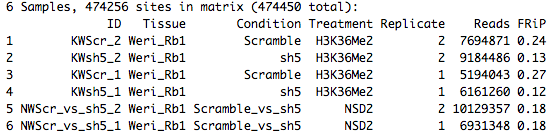
And I want to generate a venn plot like
But I got errors after I used
consKRNMWnocml <- dba.peakset(KRNMWnocml, consensus=DBA_CONDITION)
Error in dba.peakset(KRNMWnocml, consensus = DBA_CONDITION) : DBA object is already formed from a consensus peakset!
If I directly use
dba.plotVenn(KRNMWnocml,KRNMWnocml$masks$consensus)
Error in names(x) <- value : 'names' attribute [4] must be the same length as the vector [3]
If I check
KRNMWnocml$masks$consensus
NULL
If I check with capital C in $Consensus
KRNMWnocml$masks$Consensus
KWScr_2 KWsh5_2 KWScr_1 KWsh5_1 NWScr_vs_sh5_2 NWScr_vs_sh5_1 TRUE TRUE TRUE TRUE TRUE TRUE
If I used
dba.plotVenn(KRNMWnocml,KRNMWnocml$masks$Consensus)
Error in dba.plotVenn(KRNMWnocml, KRNMWnocml$masks$Consensus) : Too many peaksets in mask.
How can I get the Venn plot in plan? (I also tried "contrast" in other DBA and got a different Venn plot, but I still want to let dba.peakset() and consensus work in my hands.) Thank you very much!

Thank you so much Dr. Stark. In my last post, I simply used
KRNMWnoc <- dba.count(KRNMWnoc),dba.normalize()anddba.analyze(), beforedba.peakset(). I will switch the step ofdba.peakset()beforedba.count()for a try.Dr. Stark, I met new problem if I used
It may be because sample KWsh5_1 is from a 0-peak file. If I let the corresponding Peaks in data sheet empty, there is a warning and the DBA object could not be created.
If I replaced the empty/0-peak file into a manually created bed file containing only 1 peak with 0 signal strength, the DBA object can be smoothly created but the step of dba.peakset() got the problem as mentioned.
Could you send the output of
sessionInfo()so I can confirm what versions you are working with?Thank you Dr. Stark. Here are the information from sessionInfo()
Matrix products: default BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale: [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages: [1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages: [1] DESeq2_1.30.0 DiffBind_3.0.7 SummarizedExperiment_1.20.0 [4] Biobase_2.50.0 MatrixGenerics_1.2.0 matrixStats_0.57.0
[7] GenomicRanges_1.42.0 GenomeInfoDb_1.26.1 IRanges_2.24.0
[10] S4Vectors_0.28.0 BiocGenerics_0.36.0
loaded via a namespace (and not attached): [1] backports_1.2.0 GOstats_2.56.0 BiocFileCache_1.14.0 plyr_1.8.6
[5] GSEABase_1.52.0 splines_4.0.3 BiocParallel_1.24.1 ggplot2_3.3.2
[9] amap_0.8-18 digest_0.6.27 invgamma_1.1 GO.db_3.12.1
[13] SQUAREM_2020.5 magrittr_2.0.1 checkmate_2.0.0 memoise_1.1.0
[17] BSgenome_1.58.0 base64url_1.4 limma_3.46.0 Biostrings_2.58.0
[21] annotate_1.68.0 systemPipeR_1.24.2 askpass_1.1 bdsmatrix_1.3-4
[25] prettyunits_1.1.1 jpeg_0.1-8.1 colorspace_2.0-0 blob_1.2.1
[29] rappdirs_0.3.1 apeglm_1.12.0 ggrepel_0.8.2 dplyr_1.0.2
[33] crayon_1.3.4 RCurl_1.98-1.2 jsonlite_1.7.1 graph_1.68.0
[37] genefilter_1.72.0 brew_1.0-6 survival_3.2-7 VariantAnnotation_1.36.0 [41] glue_1.4.2 gtable_0.3.0 zlibbioc_1.36.0 XVector_0.30.0
[45] DelayedArray_0.16.0 V8_3.4.0 Rgraphviz_2.34.0 scales_1.1.1
[49] pheatmap_1.0.12 mvtnorm_1.1-1 DBI_1.1.0 edgeR_3.32.0
[53] Rcpp_1.0.5 xtable_1.8-4 progress_1.2.2 emdbook_1.3.12
[57] bit_4.0.4 rsvg_2.1 AnnotationForge_1.32.0 truncnorm_1.0-8
[61] httr_1.4.2 gplots_3.1.1 RColorBrewer_1.1-2 ellipsis_0.3.1
[65] pkgconfig_2.0.3 XML_3.99-0.5 dbplyr_2.0.0 locfit_1.5-9.4
[69] tidyselect_1.1.0 rlang_0.4.9 AnnotationDbi_1.52.0 munsell_0.5.0
[73] tools_4.0.3 generics_0.1.0 RSQLite_2.2.1 stringr_1.4.0
[77] yaml_2.2.1 bit64_4.0.5 caTools_1.18.0 purrr_0.3.4
[81] RBGL_1.66.0 xml2_1.3.2 biomaRt_2.46.0 compiler_4.0.3
[85] rstudioapi_0.13 curl_4.3 png_0.1-7 geneplotter_1.68.0
[89] tibble_3.0.4 stringi_1.5.3 GenomicFeatures_1.42.1 lattice_0.20-41
[93] Matrix_1.2-18 vctrs_0.3.5 pillar_1.4.7 lifecycle_0.2.0
[97] data.table_1.13.2 bitops_1.0-6 irlba_2.3.3 rtracklayer_1.50.0
[101] R6_2.5.0 latticeExtra_0.6-29 hwriter_1.3.2 ShortRead_1.48.0
[105] KernSmooth_2.23-18 MASS_7.3-53 gtools_3.8.2 assertthat_0.2.1
[109] openssl_1.4.3 Category_2.56.0 rjson_0.2.20 withr_2.3.0
[113] GenomicAlignments_1.26.0 batchtools_0.9.14 Rsamtools_2.6.0 GenomeInfoDbData_1.2.4
[117] hms_0.5.3 grid_4.0.3 DOT_0.1 coda_0.19-4
[121] GreyListChIP_1.22.0 ashr_2.2-47 mixsqp_0.3-43 bbmle_1.0.23.1
[125] numDeriv_2016.8-1.1
The main problem is that you are forming a consensus from two replicates where you are requiring the peaks to overlap in both replicates. As one replicate has no peaks, the consensus will be empty. There are some issues here with catching errors and giving better messages, but you will still end up with zero peaks in one of the consensus sets.
If the zero peaks is eventually what they can produce, I will reconsider if I should generate a Venn diagram about these. Thanks!
I'm checking in a version that will give a warning when there are no consensus peaks and skip generating it. It will be available in the next day or two as
DiffBind_3.0.13.It looks like you only have two replicates of each condition, and one of them generated zero peaks. Do you have any idea why you didn't get any peaks for this sample? It looks like it has a similar FrIP to the other replicate.
In this case, you can either include all the peaks from the one "good" replicate, or perhaps include the peaks in that replicate that also overlap at least one peak in one of the other conditions consensus peakset.
Here's an example using the example dataset where I've zeroed out one replicate and use a consensus consisting of the union of two consensus sets with one of the samples:
Thank you so much Dr. Stark. The 0-peak shRNA sample represented the same trends as the other replicate but missed the very tight time window which is really hard to control. Another reason may be IgG was not a good background control in this cell death condition. It is reasonable to discard the 0-peak file since the aim is to further shrink the subset of different binding sites between control and shRNA samples.
Thank you for the exemplified code lines. I previously tried sequential
dba.peakset()anddba.count()but met "Error: Can't count: some peaksets are not associated with a .bam file". Then I triedbRetrieve=TRUEindba.peakset(). But it turned out direct Granges form and the followingdba.count()gave "Error in dba.count(pKRNMWnoc) : minOverlap can not be greater than the number of peaksets [0]" How can I associate the .bam files with the DBA object after adding/replacing with "consensus" peakset? Thanks!Update: After installing the new version of Diffbind, I can smoothly run the example above. And the "associated bam" problem can be fixed by adjusting the work directory in R. Thanks! (PS: Just a note got during generating tamox.counts. 'BiocParallel' did not register default BiocParallelParam: comparison of these types is not implemented Sample: reads/Chr18_BT474_ER_2.bam125 ...)
Are you on Windows? If so that should explain the warning about BiocParallel. ( no, I see you are on MacOS...)
Yes, I am on MacOS. Interestingly, the warning about BiocParallel did not happen on my data this time but only on sample data. I already got the Venn plot from my data (before dba.count step, as the example). Thank you Dr. Stark.
And I am still thinking about these questions: 1) Since Venn diagrams are mainly about peak amounts, they are generally done at "occupancy analysis" (before dba.count) but not at "affinity analysis"(after dba.count, dba.normalize and dba.analyze). Am I right? 2)What is the best way to extract the gene symbol lists with loci and scores, from each subset of the Venn diagram?
Thanks!
Occupancy analysis basically is Venn diagrams :-) However Venns can be useful to compare differentially bound sites as well, for example overlapping the DB sites from two contrasts, or the Gain sites from one contrast with the Loss sites of another. For this you can use report-based DBA objects by setting
bDB=TRUEwhen callingdba.report()(see help page).If you assign the return value of a call to
dba.plotVenn()it will return a list of peaksets with oneGRangespeakset for each part of the Venn diagram (three for 2-way, seven for 3-way, fifteen for 4-way). You can also get these directly (without plotting) using thedba.overlap()function. TheseGRangesobjects include the loci and scores.Once you have these, annotating them can be accomplished using one of the packages developed for this purpose, such as
ChIPpeakAnnoorChIPseeker.Thank you for the very helpful instruction.