Entering edit mode

rogerbear
•
0
@user-24731
Last seen 3.2 years ago
Hi, I am trying to plot the detected expression values and average expressions of marker genes for my clustering result. I have a list of genes in genelist as the marker genes to be plotted on the y axis. But the function sort my marker gene by name each time which is not the same as their orders in my genelist. Is there a way to stop this? I also wonder if it is possible to exchange x and y axis so that the feature names are on x axis and cluster names are on y axis.
genelist
[1] "GPX3" "DCXR" "LYZ" "CD14" "CD3D" "IL7R" "CD3E"
plotDots(sce, genelist, group = "label", swap_rownames = "Symbol", max_ave = 2)
![plotDots()_result][1]
sessionInfo( )
R version 4.0.3 (2020-10-10)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur 10.16
Matrix products: default
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] scran_1.18.3 scater_1.18.3 ggplot2_3.3.3 SingleCellExperiment_1.12.0
[5] SummarizedExperiment_1.20.0 Biobase_2.50.0 GenomicRanges_1.42.0 GenomeInfoDb_1.26.2
[9] IRanges_2.24.1 S4Vectors_0.28.1 BiocGenerics_0.36.0 MatrixGenerics_1.2.1
[13] matrixStats_0.58.0 readxl_1.3.1
loaded via a namespace (and not attached):
[1] viridis_0.5.1 edgeR_3.32.1 BiocSingular_1.6.0 viridisLite_0.3.0 DelayedMatrixStats_1.12.2
[6] scuttle_1.0.4 assertthat_0.2.1 statmod_1.4.35 dqrng_0.2.1 GenomeInfoDbData_1.2.4
[11] vipor_0.4.5 cellranger_1.1.0 ggrepel_0.9.1 pillar_1.4.7 lattice_0.20-41
[16] glue_1.4.2 limma_3.46.0 beachmat_2.6.4 digest_0.6.27 XVector_0.30.0
[21] colorspace_2.0-0 cowplot_1.1.1 Matrix_1.3-2 plyr_1.8.6 pkgconfig_2.0.3
[26] zlibbioc_1.36.0 purrr_0.3.4 scales_1.1.1 BiocParallel_1.24.1 tibble_3.0.6
[31] generics_0.1.0 farver_2.0.3 ellipsis_0.3.1 withr_2.4.1 cli_2.3.0
[36] magrittr_2.0.1 crayon_1.4.0 bluster_1.0.0 beeswarm_0.2.3 tools_4.0.3
[41] lifecycle_0.2.0 stringr_1.4.0 munsell_0.5.0 locfit_1.5-9.4 DelayedArray_0.16.1
[46] irlba_2.3.3 compiler_4.0.3 rsvd_1.0.3 rlang_0.4.10 grid_4.0.3
[51] RCurl_1.98-1.2 BiocNeighbors_1.8.2 igraph_1.2.6 bitops_1.0-6 labeling_0.4.2
[56] gtable_0.3.0 DBI_1.1.1 rematch_1.0.1 reshape2_1.4.4 PCAtools_2.2.0
[61] R6_2.5.0 gridExtra_2.3 dplyr_1.0.4 stringi_1.5.3 ggbeeswarm_0.6.0
[66] Rcpp_1.0.6 vctrs_0.3.6 tidyselect_1.1.0 sparseMatrixStats_1.2.0
>
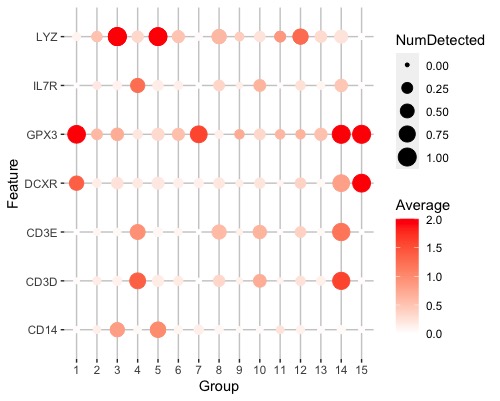



thanks for your reply, I found the scale_y_discrete really helpful, and I explored the scale_x_discrete which made my dot plot looks better.
Great! Glad I could help :)