Entering edit mode

Hello,
After reading the vignette, I decided to combine factors of interest into a single group (in this case it is Age and Genotype) rather than using an interaction term. I am interested in comparing differences between genotypes at certain ages of animals. I manually edited my matrix because there are levels without samples. When I attempt to pass in my manually edited matrix with the combined factors, I receive the error Error in solve.default(qr.R(qrx)) : 'a' (1 x 0) must be square
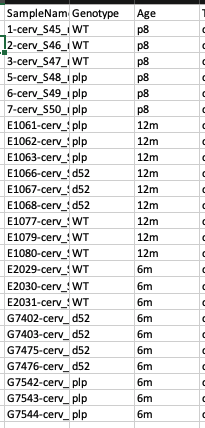
Here is what my data looks like:
ddsHTSeq <- DESeqDataSetFromHTSeqCount(sampleTable = sampletable_med,
directory = directory,
design = ~ 1)
# filter low counts
# keep only those genes that have more than 10 summed raw counts across the samples
ddsHTSeq_filtered <- ddsHTSeq[rowSums(counts(ddsHTSeq)) > 10, ]
# manually edit matrix to remove levels without samples
ddsHTSeq_filtered$group <- factor(paste0(ddsHTSeq_filtered$Genotype, ddsHTSeq_filtered$Age))
custom.matrix <- model.matrix(~ group, data = colData(ddsHTSeq_filtered))
all.zero <- apply(custom.matrix, 2, function(x) all(x==0))
idx <- which(all.zero)
custom.matrix <- custom.matrix[,-idx]
# fit statistical model
dds <- DESeq(ddsHTSeq_filtered, full = custom.matrix)
sessionInfo( )
R version 3.6.2 (2019-12-12)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] org.Mm.eg.db_3.10.0 GOstats_2.52.0 graph_1.64.0 Category_2.52.1 Matrix_1.3-2
[6] GO.db_3.10.0 AnnotationDbi_1.48.0 genefilter_1.68.0 RColorBrewer_1.1-2 gplots_3.1.1
[11] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.4 purrr_0.3.4 readr_1.4.0
[16] tidyr_1.1.2 tibble_3.0.6 tidyverse_1.3.0 ggplot2_3.3.3 pheatmap_1.0.12
[21] apeglm_1.8.0 DESeq2_1.26.0 SummarizedExperiment_1.16.1 DelayedArray_0.12.3 BiocParallel_1.20.1
[26] matrixStats_0.58.0 Biobase_2.46.0 GenomicRanges_1.38.0 GenomeInfoDb_1.22.1 IRanges_2.20.2
[31] S4Vectors_0.24.4 BiocGenerics_0.32.0
loaded via a namespace (and not attached):
[1] colorspace_2.0-0 ellipsis_0.3.1 htmlTable_2.1.0 XVector_0.26.0 base64enc_0.1-3 fs_1.5.0
[7] rstudioapi_0.13 farver_2.0.3 bit64_4.0.5 fansi_0.4.2 mvtnorm_1.1-1 lubridate_1.7.9.2
[13] xml2_1.3.2 splines_3.6.2 cachem_1.0.4 geneplotter_1.64.0 knitr_1.31 Formula_1.2-4
[19] jsonlite_1.7.2 broom_0.7.5 annotate_1.64.0 cluster_2.1.1 dbplyr_2.1.0 png_0.1-7
[25] compiler_3.6.2 httr_1.4.2 backports_1.2.1 assertthat_0.2.1 fastmap_1.1.0 cli_2.3.1
[31] htmltools_0.5.1.1 tools_3.6.2 coda_0.19-4 gtable_0.3.0 glue_1.4.2 GenomeInfoDbData_1.2.2
[37] Rcpp_1.0.6 bbmle_1.0.23.1 cellranger_1.1.0 vctrs_0.3.6 xfun_0.21 rvest_0.3.6
[43] lifecycle_1.0.0 gtools_3.8.2 XML_3.99-0.3 zlibbioc_1.32.0 MASS_7.3-53.1 scales_1.1.1
[49] hms_1.0.0 RBGL_1.62.1 memoise_2.0.0 gridExtra_2.3 emdbook_1.3.12 bdsmatrix_1.3-4
[55] rpart_4.1-15 latticeExtra_0.6-29 stringi_1.5.3 RSQLite_2.2.3 checkmate_2.0.0 caTools_1.18.1
[61] rlang_0.4.10 pkgconfig_2.0.3 bitops_1.0-6 lattice_0.20-41 labeling_0.4.2 htmlwidgets_1.5.3
[67] bit_4.0.4 tidyselect_1.1.0 AnnotationForge_1.28.0 GSEABase_1.48.0 plyr_1.8.6 magrittr_2.0.1
[73] R6_2.5.0 generics_0.1.0 Hmisc_4.4-2 DBI_1.1.1 pillar_1.5.0 haven_2.3.1
[79] foreign_0.8-75 withr_2.4.1 survival_3.2-7 RCurl_1.98-1.2 nnet_7.3-15 modelr_0.1.8
[85] crayon_1.4.1 KernSmooth_2.23-18 utf8_1.1.4 jpeg_0.1-8.1 locfit_1.5-9.4 grid_3.6.2
[91] readxl_1.3.1 data.table_1.13.6 Rgraphviz_2.30.0 blob_1.2.1 reprex_1.0.0 digest_0.6.27
[97] xtable_1.8-4 numDeriv_2016.8-1.1 munsell_0.5.0
