
Hello Everyone!
I am trying to rearrange the columns in my pheatmap, but I can't!
How do I do it?
I have bulk sequencing data were each sample preparation (Prep; 10 preps) has been subjected to two conditions (Condition; 2 conditions). I then get a heatmap of genes, but I want the columns of the heatmap to be arrange so that the columns are:
Prep1_Condition1, Prep1_Condition2, Prep2_Condition1, Prep2_Condition2, etc...
(I have a picture of this heatmap below. As you can see, the Preps don't come in pairs, and it is not like it comes with the first 10 columns being 1 condition, followed by the next 10 columns being the other condition, either!)
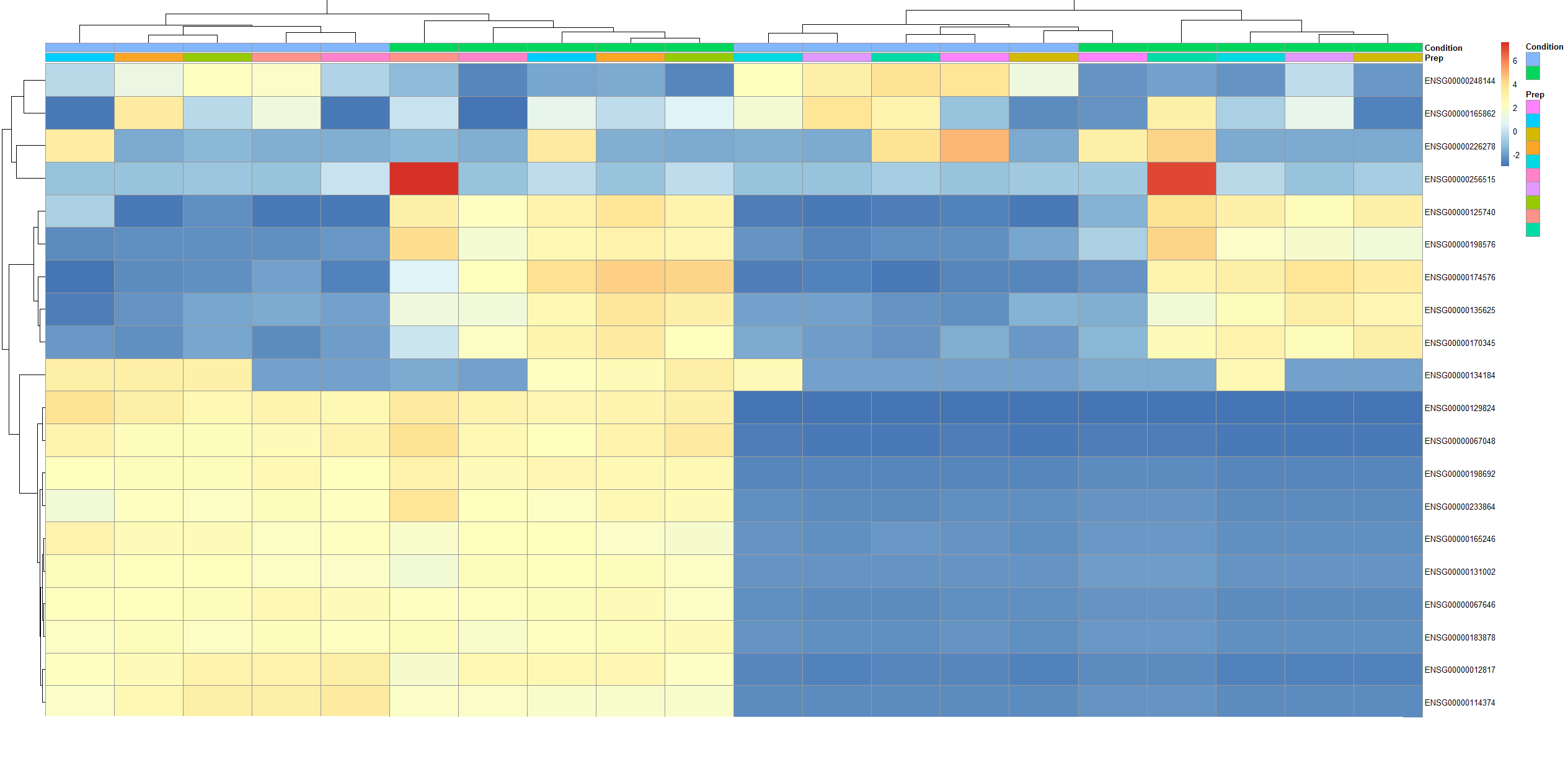
However, they are currently seemingly random. I have tried to mess around with dendsort but it hasn't done what I hoped!
The code I have used is:
library("DESeq2")
seqdata <- read.delim("Z:/featureCount.txt", stringsAsFactors = FALSE)
head(seqdata)
dim(seqdata)
countdata <- seqdata[,-(1)]
rownames(countdata) <- seqdata[,1]
# sample info (e.g. preparation ID (Prep) and experimental condition (Condition))
sampleinfo <- read.delim("Z:/SampleInfo.txt")
dds <- DESeqDataSetFromMatrix(countData = countdata,
colData = sampleinfo,
design = ~ Condition+Prep)
keep <- rowSums(counts(dds)) >= 10
dds <- dds[keep,]
dds <- DESeq(dds)
res <- results(dds)
rld <- rlog(dds)
topVarGenes <- head(order(-rowVars(assay(rld))),30)
mat <- assay(rld)[ topVarGenes, ]
mat <- mat - rowMeans(mat)
df <- as.data.frame(colData(rld)[,c("Condition","Prep")])
pheatmap(mat, annotation_col=df)
Thank you for your help!!
If you set
cluster_cols=FALSE, pheatmap will leave the order of the columns alone, and you can set it any way you like.Thank you Chris that is really helpful.
Now I have a situation whereby the columns are organised how I would like. However, it appears to be displaying the top genes that are differentially expressed across Prep as well as those differentially expressed across Condition. (See purple box in below figure.) How do I amend it so that the heatmap just shows the top differentially expressed genes with respect to Condition? This is both for up- and down-regulated genes with respect to the reference Condition. I imagine I have to change the
topVarGenesstatement.I understand this is now a slightly different question but your help with this would be really appreciated.
Just to be clear Linda, the heat map is not deciding which genes to show. You're giving it a table of numbers, and it's simply displaying them in color, and it's grouping the rows (representing genes) by similarity. If you don't want it to show the genes differentially expressed across Prep, then remove them from the table, and they won't be there to show. Does that make sense? Are you forming the input table by specifically selecting your DE gene criteria, or is this a result spit out by some program without your input?