
Hi,
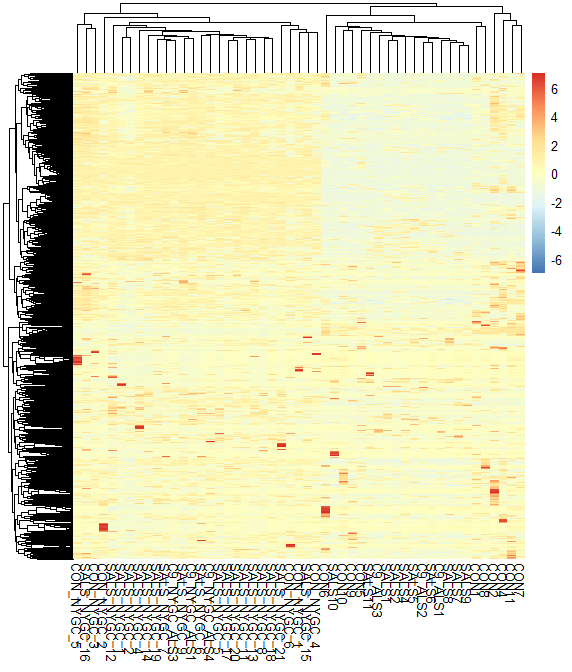
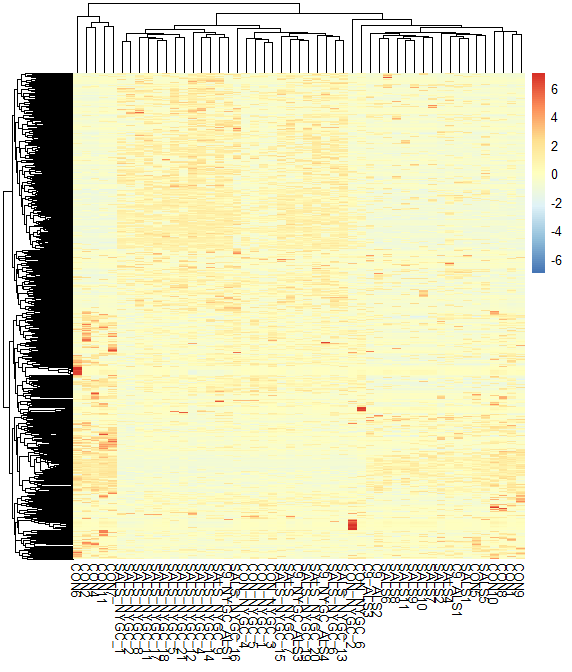
I have read blogs on how to remove batch effects in deseq2 and how to visualize it using limma voom remove batch effect PCA. I'm not sure if the batch effect was removed in my analysis. Please find the attached output, I'm skeptical because in heat map they seem to cluster separately (with respect to batches).
Analysis: 51 samples with two batches (Batch1 includes NYGC in the label and Batch2 doesn't include NYGC in the label). The two batches are rnaseq data from different sequencers.
PCA before removing batch effect
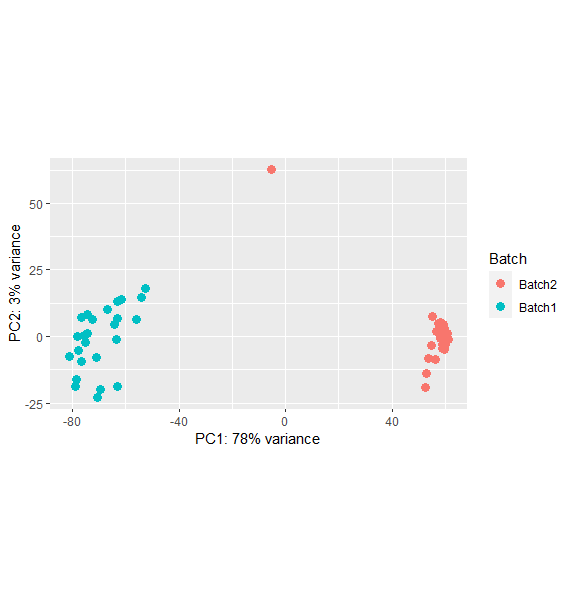
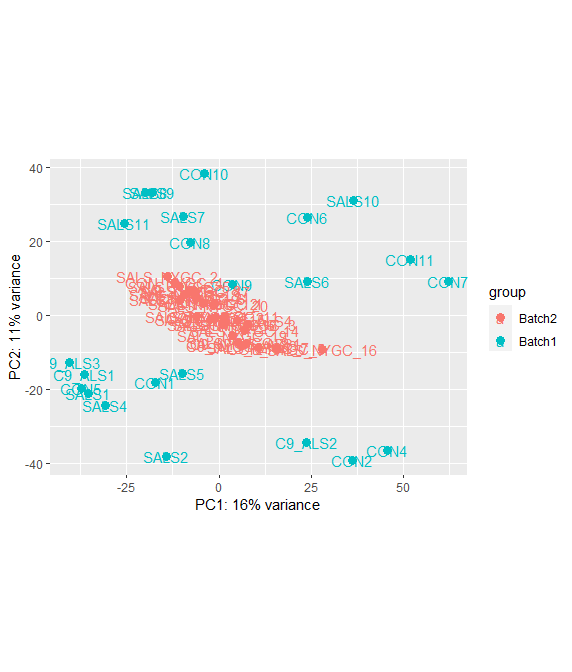
PCA after removing batch effect
Heat map with batch factor included in design formula
design =~ Gender+ageRange+Batch+Cond
Heat map without batch factor in design formula
design =~ Gender+ageRange+Cond
I'd be really great if anyone could validate my results. Thank you!
Best, Vinisha

Hi Kevin,
I've edited my post as it had an incorrect image. Kindly take a look again. Attaching the code below,
For batch effect removal I included batch in the design formula.
Ah! - now I see. Originally, it was as if literally nothing changed.
Although PC2 only accounts for 3% variation, it would be interesting to repeat everything after having remove the 'outlier' at the top of the original bi-plot. Also, is there anything else that could be confounding the differences between batch1 and batch2? Do you have statistical support for including
GenderandageRangein the design? If these are not contributing to any variation, then, by including them, you may inadvertently introduce bias that did not previously exist.I did try removing the outliers,still no luck! So I ran the analysis separately(with respect to batches) and found
GenderandageRangeto be a factor for clustering. That's the reason I included in the combined(Batch1 and Batch2) analysis. I also tried with onlyBatchin the design formula, they still clustered as per batches. Should I try a different way? Thank you for helping me out on this, Kevin!Batch1 heatmap
Batch2 heatmap
If the batch effect is inconsistent across genes, for whatever reason, then
removeBatchEffect()will struggle to adjust for this. You could instead try ComBat-Seq to remove the batch effect at the raw count stage (prior to DESeq2), if you wish; otherwise, try to explore other sources of variation via my package,PCAtools::eigencorplot(): https://github.com/kevinblighe/PCAtools#an-eigencor-plot (I have a section on how to use this with DESeq2).If all else fails, I would proceed with the analysis, possibly stratifying by batch, i.e., doing 2 separate analyses and then corroborating results across both.