
Hello,
I am adapting limma to analyse colony-based fitness data in yeast (i.e. https://en.wikipedia.org/wiki/Synthetic_genetic_array).
For this, I am using arrayWeights() to calculate weights for each plate, and duplicateCorrelation() to take care of the technical replicates (where each yeast strain is grown multiple times next to each other).
After running the linear model using lmFit(), I need to calculate the prediction variance for each fitted value. In the case of a standard linear model constructed using lm(), this could be done using predict.lm(object, se.fit=TRUE).
However, such methods are not defined for MArrayLM objects from limma. I know that the fitted values themselves can be easily calculated:
fitted <- coef(object) %*% t(design)
To calculate the variance of each fitted value (prediction variance), I have adapted the approach outlined in this post: https://stackoverflow.com/questions/39337862/linear-model-with-lm-how-to-get-prediction-variance-of-sum-of-predicted-value/39338512#39338512
I have adapted it to deal with NAs (which includes using a different QR decomposition for each model) and adjusting the way that the residual variance is calculated to incorporate weights.
In the case that there are no technical replicates, this gives the following code, where object is the matrix of fitness values being modelled, design is the design matrix, and qrList is a list of QR decompositions - one for each linear model fitted. Note that for simplicity I am only predicting values for the first gene in this code:
#Calclulate array weights
aw <- arrayWeights(object, design=design)
weights <- asMatrixWeights(aw, dim(object))
#Fit model
fit <- lmFit(object, design=design, weights=weights)
##Predict values according to model
coefs <- coef(fit)
coefs[is.na(coefs)] <- 0
predicted <- coefs %*% t(design)
predicted[is.na(object)] <- NA
##Calculate residuals
residuals <- object - predicted
#Calculate variance of fitted values for ith gene
i <- 1
#Get missing values
missing <- is.na(object[i,])
##complete variance-covariance matrix
QR <- qrList[[i]] ## qr object of fitted model
piv <- QR$pivot ## pivoting index
r <- QR$rank
B <- forwardsolve(t(QR$qr), t(design[!missing, piv]), r)
## residual variance
#sig2 <- c(crossprod(residuals[i,])) / fit$df.residual[i]
sig2 <- c(crossprod((sqrt(weights[i,]) * residuals[i,])[!missing])) / fit$df.residual[i]
## return point-wise prediction variance
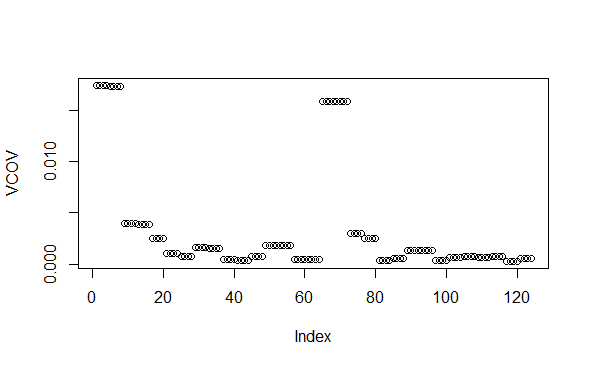
VCOV <- rep(NA, ncol(object))
VCOV[!missing] <- colSums(B ^ 2) * sig2
This code works and will give the same variance predictions as predict.lm(object, se.fit=TRUE) if I were to run a weighted linear model on the first gene using lm().
However, things become more complicated when I incorporate duplicateCorrelation() into my analysis as follows. Note that I have also used unwrapdups() to re-size object and weights, and I also re-size design:
#Calclulate array weights
aw <- arrayWeights(object, design=design)
weights <- asMatrixWeights(aw, dim(object))
#Calculate consensus correlation
dupcor <- duplicateCorrelation(myHDA, design=design, weights=aw)
#Fit model
#fit <- lmFit(object, design=design, weights=weights)
fit <- lmFit(object, design=design, correlation=dupcor$consensus.correlation, weights=weights)
#Get number of replicates and spacing
ndups <- unique(table(rownames(object)))
spacing <- 1
#Re-size to unwrap technical replicates
if(ndups > 1) {
object <- limma::unwrapdups(object, ndups=ndups, spacing=spacing)
weights <- limma::unwrapdups(weights, ndups=ndups, spacing=spacing)
design <- design %x% rep(1, ndups)
}
##Predict values according to model
coefs <- coef(fit)
coefs[is.na(coefs)] <- 0
predicted <- coefs %*% t(design)
predicted[is.na(object)] <- NA
##Calculate residuals
residuals <- object - predicted
#Calculate variance of fitted values for ith gene
i <- 1
#Get missing values
missing <- is.na(object[i,])
##complete variance-covariance matrix
QR <- qrList[[i]] ## qr object of fitted model
piv <- QR$pivot ## pivoting index
r <- QR$rank
B <- forwardsolve(t(QR$qr), t(design[!missing, piv]), r)
## residual variance
#sig2 <- c(crossprod(residuals[i,])) / fit$df.residual[i]
sig2 <- c(crossprod((sqrt(weights[i,]) * residuals[i,])[!missing])) / fit$df.residual[i]
## return point-wise prediction variance
VCOV <- rep(NA, ncol(object))
VCOV[!missing] <- colSums(B ^ 2) * sig2
This code will still provide variance estimates, but I think they are wrong. When I include duplicateCorrelation() in my pipeline, I noticed that the residual variance which I calculate, sig2, is not the same as the residual variance which comes from limma, fit$sigma[i]^2. This is not really surprising to me, although fixing it is beyond my understanding. Obviously, if I were only concerned with the residual variance, I could just use the values from limma, fit$sigma^2. However, I suspect that there may be other things going wrong because I am not taking the consensus correlation into account - perhaps calculation of the variance-covariance matrix?
So my question is this:
How do I adapt my code to account for the correlation between replicate spots when calculating the prediction variance of the fitted values?
Thank you in advance for any assistance.
Best wishes,
John

This is easy if your data doesn't have NAs. If the data has NAs, then it is considerably more complicated.
Do you really need to have NAs? I don't know of any microarray context where NAs are necessary when the data is background corrected and normalized appropriately.
Hi Gordon,
Thanks for your reply.
Yes the NAs are important. It the context of colony-based fitness screens, there are several reasons why you may have NAs.
For instance, we array collections of deletion mutants in 384-well format, meaning that each collection is spread over around a dozen agar plates. You can consider the plates as analagous to subarrays - i.e.
printtipWeights(). It's reasonably common for a single plate within a collection to fail - perhaps becasue of a microbial contamination on the plate, or because of a pinning error on the robot. So we deal with this by setting the entire plate to NA.Another important source of NAs comes from the batches. The batch effects look a little different in colony-based fitness screens. There are some mutants which do not wake up from the cryostocks very efficiently, and hence some mutants are missing in each screen. Importantly, the mutants which are missing differ on a batch-by-batch basis. This obviously creates problems in detecting fitness differences for strains which are missing in some batches. Consider a mutant which is lethal in when grown on a drug. In batch 1, where the mutant is present, the normalised colony sizes would be ~1 on the control plate and 0 on the drug. If this mutant were missing in batch 2, the normalised colony sizes would be 0 and 0 - suggesting no effect. We deal with this by analysing which mutants are missing on the control plate for each batch, and then setting the missing mutants as NA on a batch-by-batch basis.
So these NAs reflect important quality control steps which we have developed to analyse colony-based fitness data.
What is the problem caused by including NAs in the analysis? From what I understand is happening inside
gls.series(), it's just a case of dropping the NAs before callinglm.fit().Best wishes,
John
If you don't have NAs then limma stores all the information you need as part of the MArrayLM object and getting the prediction variances is a couple of lines of code. If you do have NAs, then you have to reproduce and modify the entire limma lmFit code pipeline from scratch, because limma does not store genewise QRs.
I have already written a modified
gls.series()to save the genewise QRs. And I know how to get prediction variances in the case that there are no technical replicates. Does usingduplicateCorrelation()change this? I know that the residual variance needs to be calculated differently in the case that there are technical replicates. Are there any other changes which I need to implement? Such as changing the way the variance-covariance matrix is calculated, or the way the prediciton variance is calculated from the variance-covariance matrix?I am concerned that you seem to have NA coefficients and fitted values as well as NA observations. That would imply that your linear model is not always estimable, which really puts in doubt the interpretation of the linear model on those cases.
Yes you are correct, the linear model is not always estimable. But as per my reply above, it is sometimes things like batch effects which we cannot estimate - which is not a problem if an entire batch is missing. But I agree that there may also be instances where coefficients of interest may be misleading. I hadn't considered this, thanks for pointing this out.
You certainly don't need to set predicted values to NA just because the corresponding data value was. If the coefficients are not NA, then all the fitted values are estimable.