Hi,
You already received an answer on Biostars: https://www.biostars.org/p/9473246/#9473248
The different annotation databases do not overlap perfectly. Each [database] includes transcripts and genes based on different rules. There are actually many previous questions on this topic, and even publications. It is not strictly an issue with the TCGAbiolinks package.
Let's see if we can find out more about the two circled, i.e., ENSG00000281904 and ENSG00000281920:
genes <- c('ENSG00000281904','ENSG00000281920')
1, via org.Hs.eg.db
require(org.Hs.eg.db)
mapIds(
org.Hs.eg.db,
keys = genes,
column = 'SYMBOL',
keytype = 'ENSEMBL')
Error in .testForValidKeys(x, keys, keytype, fks) :
None of the keys entered are valid keys for 'ENSEMBL'. Please use the keys method to see a listing of valid arguments.
Not there.
2, via biomaRt
require(biomaRt)
ensembl <- useMart('ensembl', dataset = 'hsapiens_gene_ensembl')
annot <- getBM(
attributes = c(
'hgnc_symbol',
'external_gene_name',
'ensembl_gene_id',
'entrezgene_id',
'gene_biotype'),
filters = 'ensembl_gene_id',
values = genes,
mart = ensembl)
annot <- merge(
x = as.data.frame(genes),
y = annot,
by.y = 'ensembl_gene_id',
all.x = T,
by.x = 'genes')
annot
genes hgnc_symbol external_gene_name entrezgene_id gene_biotype
1 ENSG00000281904 NA NA NA lncRNA
2 ENSG00000281920 NA NA NA lncRNA
Nothing there either, but at least that we can see that these are long non-coding RNAs.
Kevin

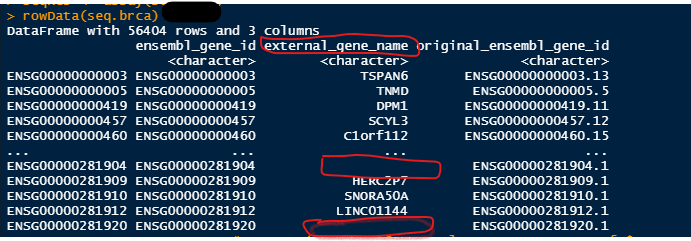

Thank you Kevin, So do you mean that I can neglect them for my analysis? Actually, when I used the "gencode.gene.info.v22.csv" file from TCGA, it has assigned some name to them (highlighted part in the first picture attached).
But on the other hand, my friend get the exact name of the genes one year ago by "gencode.gene.info.v22.csv", but they are not the same, I mean they have aliases. for example;
RP11-418H16.1 = AC007389.5
CH17-132F21.5= AC233263.6
So I'm wondering how can I get the same gene names such as "AC007389.5" instead of "RP11-418H16.1" as I mentioned above. ?