
Hi,
I am performing differential expression on gene expression between different time points- time point 0 vs. time points 0.5, 1 and 3.
For a specific gene, I have the following expression profile (this is normalized reads):
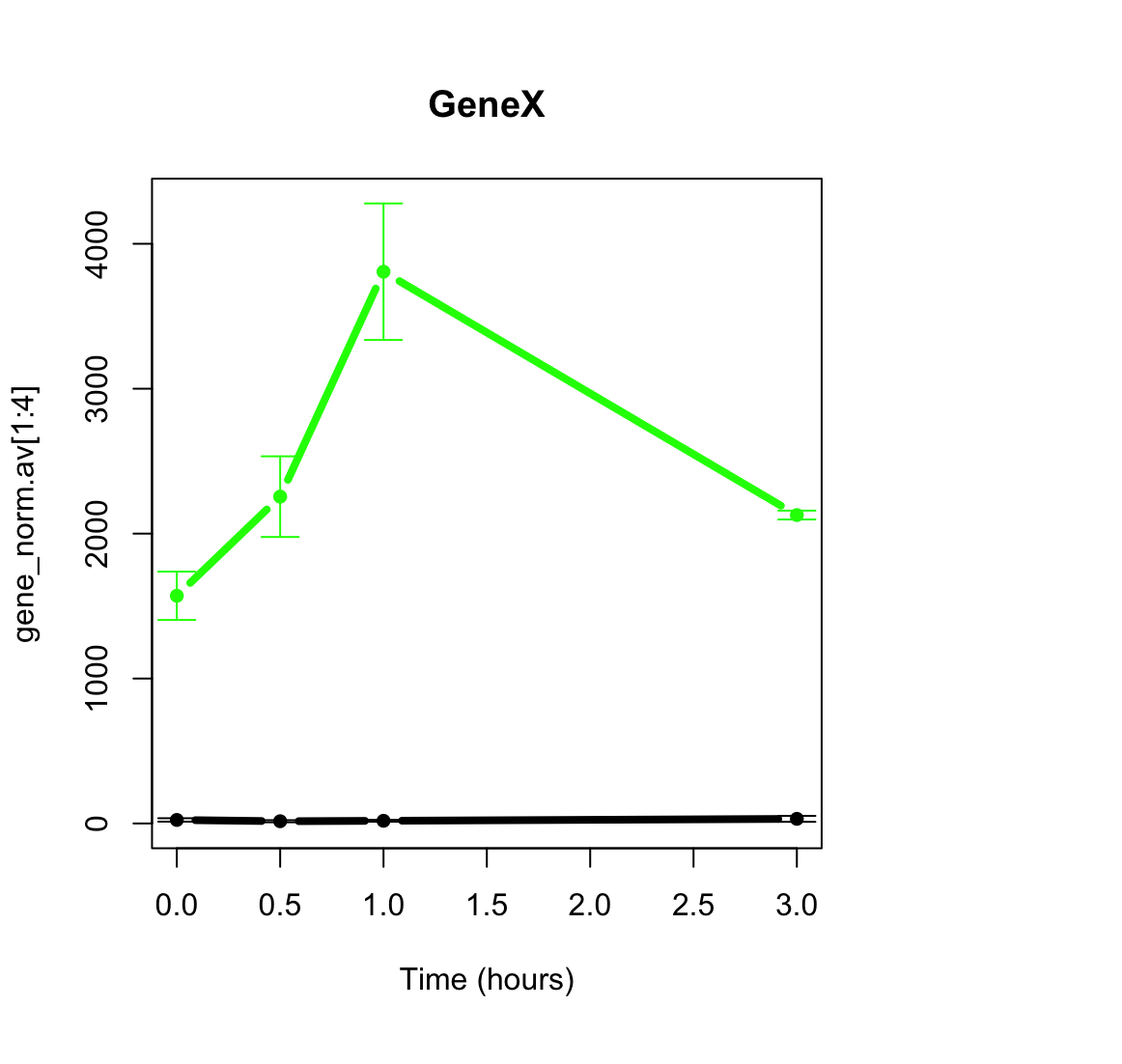
It seems in the plot like there is a significant difference between time points 0 and 1. Actually, this is a gene which I know is induced in this paradigm from many past experiments. However, DESeq does not find the comparison significant:
baseMean log2FoldChange lfcSE stat pvalue padj
GeneX 1230.466 1.277551 1.164463 1.097116 0.2725906 0.8389563
The individual values of time point 0 are:
tp0.1 tp0.2 tp0.3
1622.129 1260.146 1831.165
and for 1:
tp1.1 tp1.2 tp1.3
2878.453 4137.590 4406.023
If I perform a simple t-test on the two time points, I get p-value = 0.0304.
My question is- what might be the reason that this comparison is coming up as insignificant in the DESeq analysis? Thanks a lot!

Hey Michael, thanks so much for your reply. I've uploaded the raw reads and colData to:
https://drive.google.com/file/d/1ryg73rZbsvVjIwp4drhF2iONVTHWDeAY/view?usp=sharing
https://drive.google.com/file/d/1JsnOpEP2D7JkTBsN7kcAKUkyq-q1Hth_/view?usp=sharing
The code is:
Two recommendations, the two cell types are very different, for a gene like GeneX, it's not even expressed in the other cell type. So one is to split these within cell type comparisons by making two
ddsobjects. The second is to remove thousands of unexpressed genes once you split into the two datasets (either way I would recommend to do this for speed).You're better off modeling like this:
Hi Michael,
A follow up to the above question: I've performed the comparison for each cell type in a different dds, as you suggested. Next, I wish to plot the timecourse of expression for both cell types on the same graph. However, now the normalized counts are no longer from a normalization of all samples together (rather, each dds of each cell types was normalized on its own). What is the correct way to compare and plot the data in such a case?
Thanks again!
I'd recommend to compute
vstof the entire dataset and use those values for plotting. These are ~ log2 counts for large counts but stabilized as the counts approach 0.