
Hello!
I have run into a result I cannot explain with DESeq2 and would greatly appreciate an explanation!
I am running DESeq2 and for 197 samples for two groups of comparison. I am not currently interested in interaction terms. My code looks as follows
dds <- DESeqDataSetFromMatrix(counts_ent_T,design_df , ~ param1+param2)
filter<-rowSums(counts(dds)>=10)>=10
dds<-dds[filter,]
dds<-DESeq(dds,parallel=T)
dds<-dds [which(mcols(dds)$betaConv), ]
and design_df looks as follows:
> design_df [1:6,]
param1 param2
Sample1 bulk bulk
Sample2 bulk bulk
Sample3 bulk bulk
Sample4 bulk bulk
Sample5 outlier outlier
Sample6 bulk bulk
where param1 has 30 "outlier"s and 167 "bulk"s, and param2 has 28 "outlier"s and 169 "bulk"s. Except for 4 samples param1 and param2 are the same.
The problem is in the results. As an example, I have looked at "Gene1":
res1<-results(dds,name="param1_outlier_vs_bulk")
res2<-results(dds,name="param2_outlier_vs_bulk")
> res1["Gene1",]
log2 fold change (MLE): param1 outlier vs bulk
Wald test p-value: param1 outlier vs bulk
DataFrame with 1 row and 6 columns
baseMean log2FoldChange lfcSE stat pvalue padj
<numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
Gene1 255.719 -12.7665 2.55447 -4.99771 5.80142e-07 0.000895952
> res2["Gene1",]
log2 fold change (MLE): param2 outlier vs bulk
Wald test p-value: param2 outlier vs bulk
DataFrame with 1 row and 6 columns
baseMean log2FoldChange lfcSE stat pvalue padj
<numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
Gene1 255.719 13.0614 2.61151 5.00145 5.69003e-07 0.000906422
As you can see, the log2FoldChange is exactly opposite, while I didn't swap factor levels in the comparisons (comparing outlier vs bulk in both params). Moreover, plotcounts look as follows:
p1<-plotCounts(dds, gene=gene1, intgroup="param1", returnData=T)
p11<-ggplot(p1, aes(x=param1, y=count)) +
geom_point(position=position_jitter(w=0.1,h=0)) +ggtitle("Gene1")+
scale_y_log10(breaks=c(25,100,400))
ggsave(p11, file="plotcounts_param1_default.pdf")
p2<-plotCounts(dds, gene=gene1, intgroup="param2", returnData=T)
p22<-ggplot(p2, aes(x=param2, y=count)) +
geom_point(position=position_jitter(w=0.1,h=0))+ggtitle("Gene1")+
scale_y_log10(breaks=c(25,100,400))
ggsave(p22, file="plotcounts_param2_default.pdf")
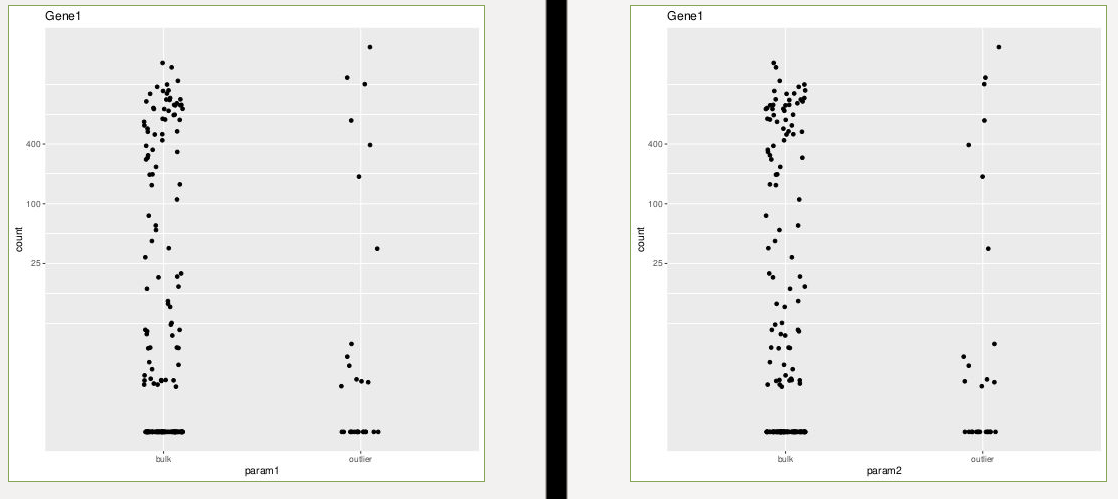
So why are the log2FoldChanges exactly opposite? Is it really just the 2 samples with 0s that the outlier group in param1 gains?
Any help would be greatly appreciated.
Rita
sessionInfo()
R version 4.0.2 (2020-06-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.3 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=de_DE.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=de_DE.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=de_DE.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=de_DE.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] BiocParallel_1.24.1 purrr_0.3.4
[3] ggplot2_3.3.3 dplyr_1.0.4
[5] plyr_1.8.6 DESeq2_1.30.1
[7] SummarizedExperiment_1.20.0 Biobase_2.50.0
[9] MatrixGenerics_1.2.1 matrixStats_0.58.0
[11] GenomicRanges_1.42.0 GenomeInfoDb_1.26.2
[13] IRanges_2.24.1 S4Vectors_0.28.1
[15] BiocGenerics_0.36.0 data.table_1.14.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.6 locfit_1.5-9.4 lattice_0.20-41
[4] snow_0.4-3 digest_0.6.27 assertthat_0.2.1
[7] utf8_1.1.4 R6_2.5.0 RSQLite_2.2.3
[10] httr_1.4.2 pillar_1.5.0 zlibbioc_1.36.0
[13] rlang_0.4.10 annotate_1.68.0 blob_1.2.1
[16] Matrix_1.2-18 labeling_0.4.2 splines_4.0.2
[19] geneplotter_1.68.0 RCurl_1.98-1.2 bit_4.0.4
[22] munsell_0.5.0 DelayedArray_0.16.2 compiler_4.0.2
[25] pkgconfig_2.0.3 tcltk_4.0.2 tidyselect_1.1.0
[28] tibble_3.1.0 GenomeInfoDbData_1.2.4 XML_3.99-0.5
[31] fansi_0.4.2 crayon_1.4.1 withr_2.4.1
[34] bitops_1.0-6 grid_4.0.2 xtable_1.8-4
[37] gtable_0.3.0 lifecycle_1.0.0 DBI_1.1.1
[40] magrittr_2.0.1 scales_1.1.1 cachem_1.0.4
[43] farver_2.1.0 XVector_0.30.0 genefilter_1.72.1
[46] ellipsis_0.3.1 vctrs_0.3.6 generics_0.1.0
[49] RColorBrewer_1.1-2 tools_4.0.2 bit64_4.0.5
[52] glue_1.4.2 fastmap_1.1.0 survival_3.1-12
[55] AnnotationDbi_1.52.0 colorspace_2.0-0 memoise_2.0.0

Thank you, Michael. After your response, I found that running DESeq for each parameter separately seems to solve my problem and gives sensible results. I will just proceed with that!