
Hello!
First of all, thank you for the great package and the excellent documentation that supports it, much appreciated!
Sadly, I could not find an answer to my problem, so I wanted to ask here.
I have two different bulk RNA-seq datasets, one obtained from TCGA using the TCGAbiolinks package, and one that I acquired after communicating with the authors. They are both from the same cancer type, but the TCGA consists of patient samples exclusively from later stages and the other exclusively from earlier stages.
Following the TCGAbiolinks vignette, I got a SummarizedExperiment object that is ready to proceed with the DESeq2 workflow. The raw counts are HTSeq quantified.
The second dataset, was given to me as an excel file of raw counts quantified with Salmon so I had to bring it to the appropriate format:
#Assuming all the relevant packages are loaded
raw.salmon <- readxl::read_excel(file.choose())
clin.salmon <- read_excel(file.choose()) #Relevant clinical data to be used as metadata
raw.salmon <- raw.salmon %>% remove_rownames() %>% column_to_rownames(var = 'ensembl ID')
raw.salmon <- raw.salmon %>% mutate_if(is.numeric, round) #Salmon raw counts included decimals, so I rounded them. I don't believe meaningful information is lost.
raw.salmon <- raw.salmon %>% mutate_if(is.numeric, as.double) # This was the type of the counts from the other SummarizedExperiment object from TCGAbiolinks
raw.salmon <- as.matrix(raw.salmon)
raw.salmon.se <- SummarizedExperiment(raw.salmon)
assayNames(raw.salmon.se) <- 'counts'
raw.salmon.se$Stage <- clin.salmon$Stage
Then I proceeded to "merge" the two together, so I could do DE analysis based on stage. The code is a bit messy here since I had saved the two Summarized experiments and came back to them at a different day so you see me going back and forth to matrices, data.frames etc...
#raw.htseq.se is the SummarizedExperiment from TCGAbiolinks
# Extract the counts into matrices
htseq.matrix <- raw.htseq.se@assays@data@listData[["HTSeq - Counts"]]
salmon.matrix <- raw.salmon.se@assays@data@listData[["counts"]]
#Add rownames and colnames to htseq.matrix to be the same as salmon
colnames(htseq.matrix) <- rownames(colData(htseq.salmon.se))
names.htseq<- rowData(htseq.salmon.se)
rownames(htseq.matrix) <- names.htseq$ensembl_gene_id
#Turn them to data frames for easier manipulation
htseq.matrix <- as.data.frame(htseq.matrix) # I know... data.frames names matrices...
salmon.matrix <- as.data.frame(salmon.matrix)
#Change the rownames into a column as a reference for merging
htseq.matrix <- htseq.matrix %>% rownames_to_column('Ensembl_ID')
salmon.matrix <- salmon.matrix %>% rownames_to_column('Ensembl_ID')
combined.counts <- merge(salmon.matrix, htseq.matrix, by = 'Ensembl_ID')
combined.counts <- combined.counts %>% column_to_rownames('Ensembl_ID')
combined.counts <- combined.counts %>% mutate_if(is.numeric, as.double)
combined.counts <- as.matrix(combined.counts)
#Turn into a SummarizedExperiment for DE analysis with DESeq2
combined.counts.se <- SummarizedExperiment(combined.counts.se)
assayNames(combined.counts.se) <- 'counts'
combined.counts.se$Stage <- all.clin$Stage # This is a table I made that had the appropriate stage metadata for both datasets.
After this, I could turn the combined SummarizedExperiment object into a DESeqDataSet and proceed to DE analysis using design = ~ Stage without a problem and I got plenty of results but, are they correct?
Is it appropriate to run such a workflow with raw data that have been called differently?
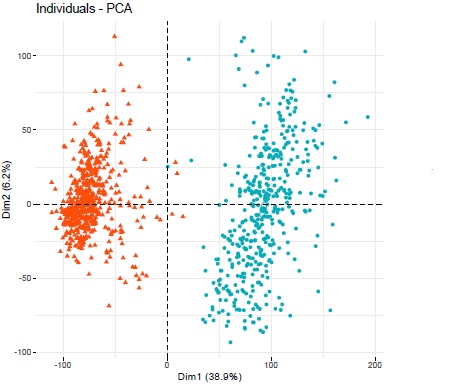
Running a PCA grouping the individuals based on stage (after transforming the counts with vst and blind = TRUE) gave me two big clusters, one for the early stage (orange) and one for the later stage (blue) which also happens to be the two different datasets respectively. Is this whole analysis just a big batch effect?
Any opinions are more than welcome!


That is what I was afraid of and wanted to make sure before spending any more time on it. Will have to think about something else then.
Thanks again!