
Hello!
We are using DESeq2 to analyse RNAseq data from an experiment and are running into convergence errors that we are unable to solve.
We are not sure what is causing these errors; and would preferably solve them rather than dropping the genes with the error from the results.
We already tried the advice given here: DESeq2 Error: rows did not converge in beta, labelled in mcols(object)$betaConv. Use larger maxit argument with nbinomWaldTest
We prefilter for genes expressed with count>10 in 2 of the 3 replicates at least on one occassion (timepoint/treatment combination); which is about as strict as we want to go. And we increased the maximum iterations to maxit=10,000
Leaving us still with "261 rows did not converge in beta" out of 25 480 genes in total.
Looking at the expression patterns of the genes that don't converge, many seem to be presence/absence genes: we observe many zero counts, and for the samples where these genes are expressed, the expression range is similar (see figures included below).
Anyone have an idea how to deal with these genes?
Thanks! Natalie
Heat map z-scores of raw counts for 261 genes with convergence errors, all 48 samples shown
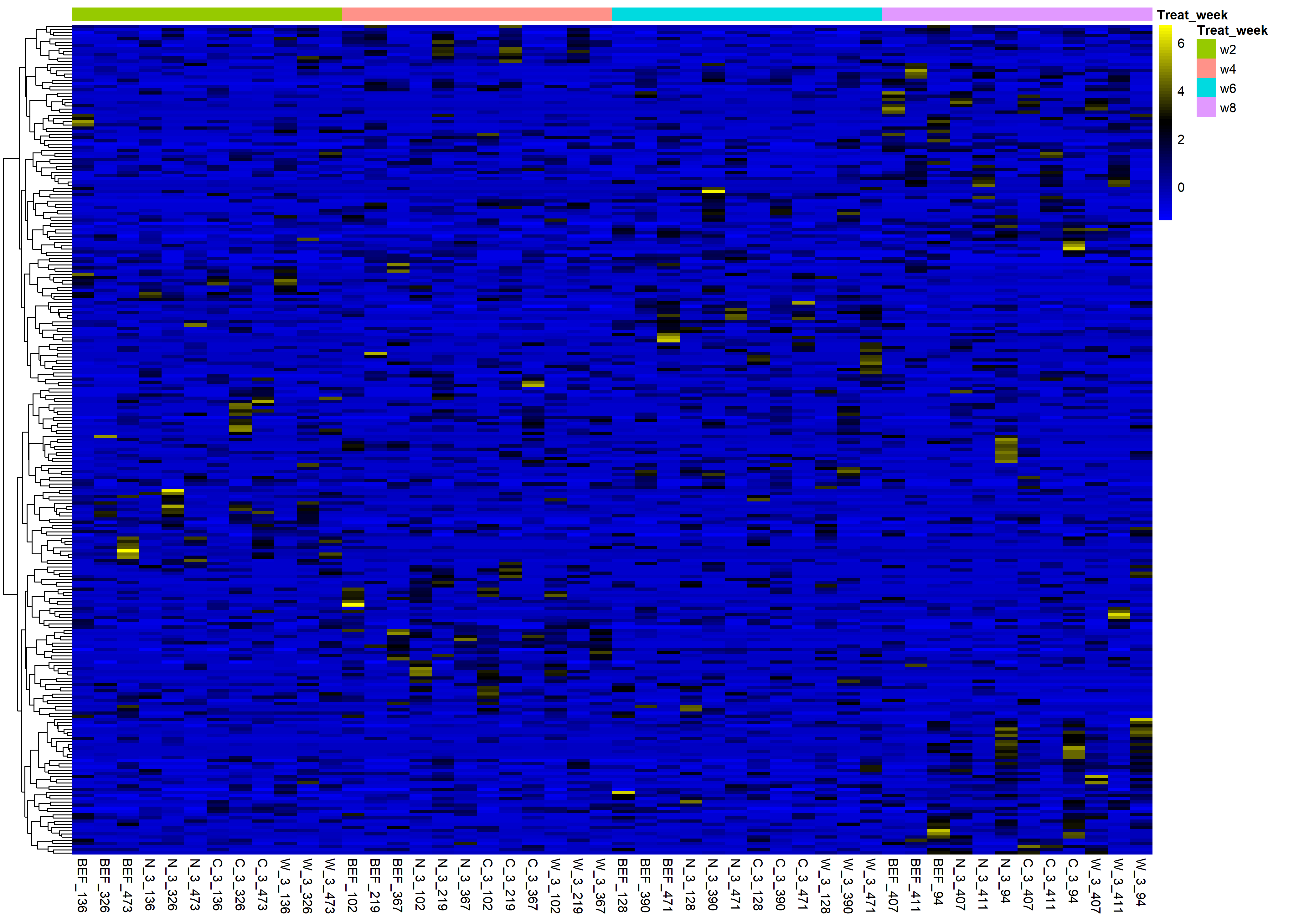
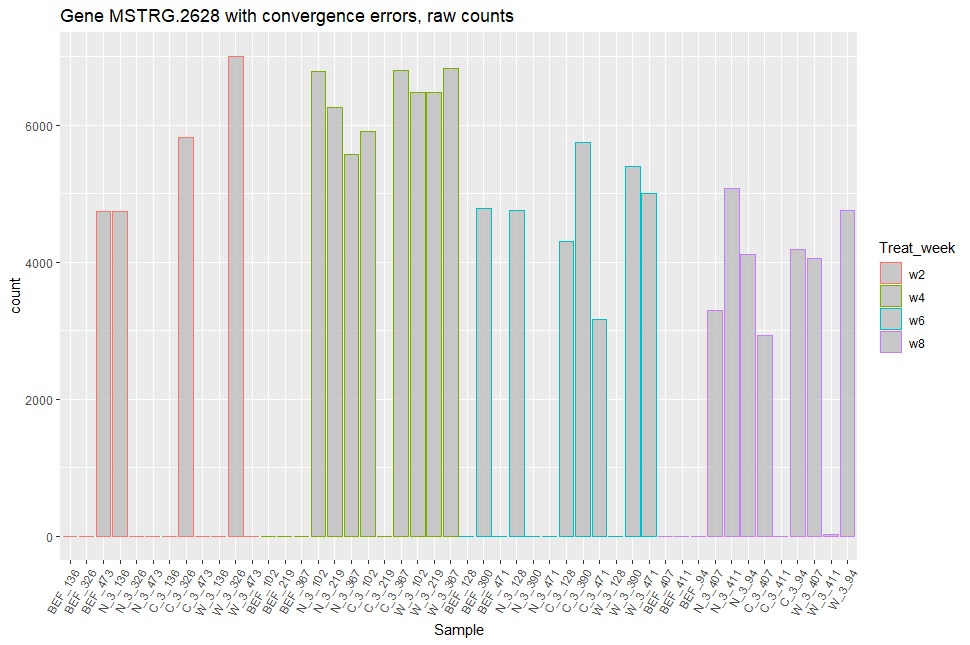
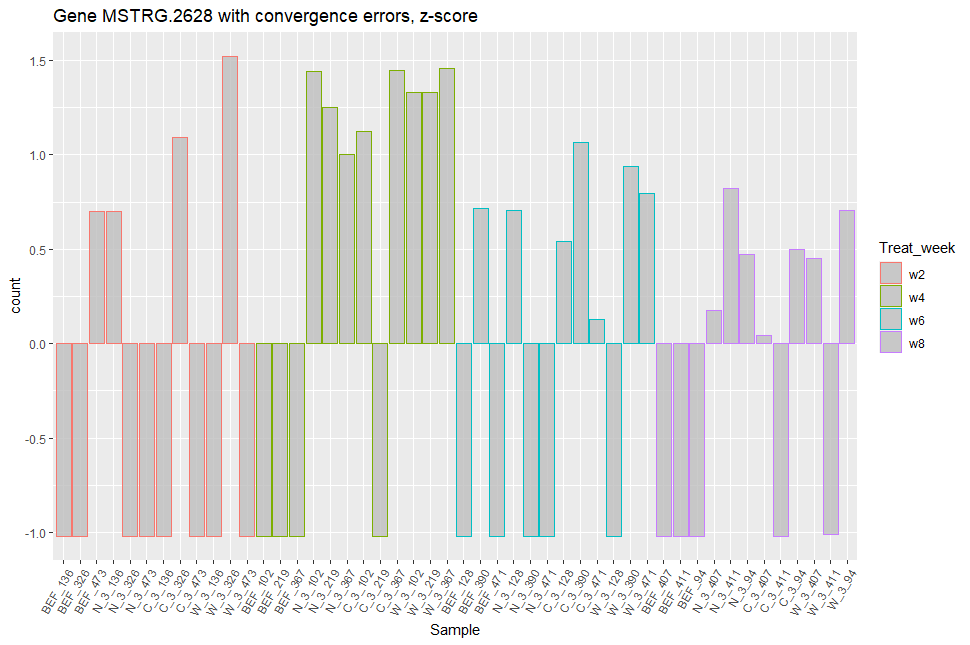
Code for construction and running DESeq2:
> # Design for Treatment effects within and between weeks, all weeks together
> # Correct for individual effects nested in group with Tube.n
> design <- ~ -1 + Treat_week + Treat_week:Tube.n + Treatment + Treatment:Treat_week
> dds <- DESeqDataSetFromMatrix(countData = counts,
colData= sampleinfo,
design = design)
> dds@colData
DataFrame with 48 rows and 6 columns
Sample Treat_week Tube Tube.n Treatment sizeFactor
<character> <factor> <factor> <factor> <factor> <numeric>
BEF_136 w2 136 1 BEF 0.9003222
N_3_136 w2 136 1 N 1.0020815
C_3_136 w2 136 1 C 0.9995351
W_3_136 w2 136 1 W 1.0937525
BEF_326 w2 326 2 BEF 0.7327493
N_3_326 w2 326 2 N 1.0197463
C_3_326 w2 326 2 C 1.0153445
W_3_326 w2 326 2 W 1.1075391
BEF_473 w2 473 3 BEF 0.9488591
N_3_473 w2 473 3 N 0.6653621
C_3_473 w2 473 3 C 0.8698424
W_3_473 w2 473 3 W 0.9524100
BEF_102 w4 102 1 BEF 1.0311267
N_3_102 w4 102 1 N 1.0104705
C_3_102 w4 102 1 C 1.0675697
W_3_102 w4 102 1 W 1.0146415
BEF_219 w4 219 2 BEF 0.9445711
N_3_219 w4 219 2 N 1.1474465
C_3_219 w4 219 2 C 0.9889870
W_3_219 w4 219 2 W 1.1061815
BEF_367 w4 367 3 BEF 0.9656871
N_3_367 w4 367 3 N 0.8936870
C_3_367 w4 367 3 C 0.9712031
W_3_367 w4 367 3 W 0.9428335
BEF_128 w6 128 1 BEF 0.9951708
N_3_128 w6 128 1 N 1.0238084
C_3_128 w6 128 1 C 1.0812198
W_3_128 w6 128 1 W 1.0628506
BEF_390 w6 390 2 BEF 0.9483895
N_3_390 w6 390 2 N 1.0471350
C_3_390 w6 390 2 C 1.0149936
W_3_390 w6 390 2 W 0.9980588
BEF_471 w6 471 3 BEF 0.9381325
N_3_471 w6 471 3 N 1.0343588
C_3_471 w6 471 3 C 0.9019616
W_3_471 w6 471 3 W 1.1463979
BEF_407 w8 407 1 BEF 1.0869852
N_3_407 w8 407 1 N 1.1773927
C_3_407 w8 407 1 C 1.0102222
W_3_407 w8 407 1 W 1.1481354
BEF_411 w8 411 2 BEF 0.9814388
N_3_411 w8 411 2 N 1.1090623
C_3_411 w8 411 2 C 1.0564870
W_3_411 w8 411 2 W 1.0433508
BEF_94 w8 94 3 BEF 1.0275384
N_3_94 w8 94 3 N 1.3217692
C_3_94 w8 94 3 C 1.2059190
W_3_94 w8 94 3 W 1.2727506
> # Prefilter gene set
> dds.filt <- dds[rownames(dds) %in% filtwhich.n2,] # only keep genes expressed with count>10 in >=2 replicates at least in one timepoint/treatment combi
> dim(dds)
[1] 29113 48
> dim(dds.filt)
[1] 25480 48
> # Run model
> dds.3h <- estimateSizeFactors(dds.filt)
> dds.3h <- estimateDispersions(dds.3h)
> dds.3h <- nbinomWaldTest(dds.3h, maxit=10000) #default maxit=100
> table(mcols(dds.3h)$betaConv, useNA="always")
FALSE TRUE <NA>
261 25217 2
> sessionInfo()
R version 4.1.0 (2021-05-18)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 17763)
Matrix products: default
locale:
[1] LC_COLLATE=Dutch_Netherlands.1252 LC_CTYPE=Dutch_Netherlands.1252 LC_MONETARY=Dutch_Netherlands.1252 LC_NUMERIC=C
[5] LC_TIME=Dutch_Netherlands.1252
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] pheatmap_1.0.12 DESeq2_1.32.0 SummarizedExperiment_1.22.0 Biobase_2.52.0 MatrixGenerics_1.4.2
[6] matrixStats_0.60.0 GenomicRanges_1.44.0 GenomeInfoDb_1.28.1 IRanges_2.26.0 S4Vectors_0.30.0
[11] BiocGenerics_0.38.0 PCAtools_2.4.0 ggrepel_0.9.1 forcats_0.5.1 stringr_1.4.0
[16] dplyr_1.0.7 purrr_0.3.4 readr_2.0.1 tidyr_1.1.3 tibble_3.1.2
[21] ggplot2_3.3.5 tidyverse_1.3.1
loaded via a namespace (and not attached):
[1] bitops_1.0-7 fs_1.5.0 lubridate_1.7.10 bit64_4.0.5 RColorBrewer_1.1-2 httr_1.4.2
[7] tools_4.1.0 backports_1.2.1 irlba_2.3.3 utf8_1.2.1 R6_2.5.1 DBI_1.1.1
[13] colorspace_2.0-2 withr_2.4.2 tidyselect_1.1.1 bit_4.0.4 compiler_4.1.0 fdrtool_1.2.16
[19] cli_3.0.1 rvest_1.0.1 xml2_1.3.2 DelayedArray_0.18.0 scales_1.1.1 genefilter_1.74.0
[25] XVector_0.32.0 pkgconfig_2.0.3 sparseMatrixStats_1.4.2 dbplyr_2.1.1 fastmap_1.1.0 rlang_0.4.11
[31] GlobalOptions_0.1.2 readxl_1.3.1 rstudioapi_0.13 RSQLite_2.2.7 DelayedMatrixStats_1.14.2 shape_1.4.6
[37] ggVennDiagram_1.1.4 generics_0.1.0 jsonlite_1.7.2 BiocParallel_1.26.1 BiocSingular_1.8.1 RCurl_1.98-1.3
[43] magrittr_2.0.1 GenomeInfoDbData_1.2.6 Matrix_1.3-3 Rcpp_1.0.7 munsell_0.5.0 fansi_0.5.0
[49] lifecycle_1.0.0 stringi_1.6.2 zlibbioc_1.38.0 plyr_1.8.6 grid_4.1.0 blob_1.2.2
[55] dqrng_0.3.0 crayon_1.4.1 lattice_0.20-44 beachmat_2.8.1 cowplot_1.1.1 Biostrings_2.60.1
[61] haven_2.4.3 splines_4.1.0 annotate_1.70.0 circlize_0.4.13 hms_1.1.0 KEGGREST_1.32.0
[67] locfit_1.5-9.4 pillar_1.6.2 reshape2_1.4.4 ScaledMatrix_1.0.0 geneplotter_1.70.0 reprex_2.0.1
[73] XML_3.99-0.6 glue_1.4.2 data.table_1.14.0 modelr_0.1.8 png_0.1-7 vctrs_0.3.8
[79] tzdb_0.1.2 cellranger_1.1.0 gtable_0.3.0 assertthat_0.2.1 cachem_1.0.5 rsvd_1.0.5
[85] xtable_1.8-4 broom_0.7.9 survival_3.2-11 RVenn_1.1.0 AnnotationDbi_1.54.1 memoise_2.0.0
[91] ellipsis_0.3.2
