
Hello everyone,
I am doing WGCNA on RNA-Seq data, where samples of 3 different conditions are taken from the same mice. More than one sample was taken for each condition from the same mice ( as different regions). I did vst and removed batch effect. I have two main questions: 1- How to remove low expression genes, knowing that the over all pattern of expression values is low, This is for both differential analysis and WGCNA Note: Fractions because I averaged samples taken from the same condition and same mice. These are the raw counts.
0610009B22Rik 8.5 18.5 21 19.33333333 13 14 7.5 5 6.666666667 23 3 6 19 15 24.33333333 16 21.5 9 0610010K14Rik 16 17.5 23.33333333 14 12 7.5 9 3 10.33333333 15 5.5 1.5 21.5 20.5 22 15.66666667 16.5 3.5 0610012G03Rik 16.5 24.5 24 18 17.5 14.5 6 5 12.66666667 30 6.5 5.5 17 15 23.66666667 16.66666667 37 6.5 0610030E20Rik 13.5 20 18.66666667 16 7.5 4.5 7 6 9 22 4 2.5 16.5 16 20.66666667 13.33333333 18 4.5
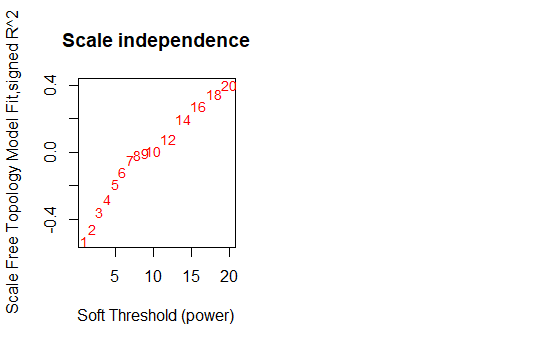
2- I am planning to do WGCNA using all samples without doing average. How to select the sft from these two figures? scale independence mean connectivity according to mean connectivity I would choose 8-9 but the scale independence is confusing and I don't understand why the curve started to rise again after 9?
Many thanks
Code should be placed in three backticks as shown below
# include your problematic code here with any corresponding output
# please also include the results of running the following in an R session
sessionInfo( )