
Hello everyone,
My dataset consists of 16 samples for which 784 genes (730 endogenous, 40 housekeeping, 8 negative controls and 6 positive controls) were quantified with NanoString nCounter technology in three different and known batches. My aim is to perform diferential expression between two groups of 10 and 6 samples.
After reading several posts about the use of DESeq2 on NanoString data, and following the tutorials and code provided in the GitHub linked in the publication An approach for normalization and quality control for NanoString RNA expression data, I have reached and impasse in my analysis. The housekeeping genes, which are supossed to be estable across conditions, do not meet such requirement. I have tested this using the following code on the raw data:
#### CHECK IF HK ARE ASSOCIATED WITH PRIMARY PHENO
hk_raw = raw[cIdx,]
pval = vector(length = nrow(hk_raw))
for (i in 1:nrow(hk_raw)){
reg = glm.nb(as.numeric(hk_raw[i,]) ~ as.factor(pData$Group))
pval[i] = coef(summary(reg))[2,4]
}
This results in 38 p-values under .05, which points to an association between expression of these so called housekeeping genes and the phenotype.
Following the pipeline, I estimated RUVg factors (using all of the 40 housekeeping genes) and used the following code to create the DESeq object:
dds <- DESeqDataSetFromMatrix(countData = counts(set)[c(1:730,745:784),],
colData = pData(set),
design = ~ W_1 + Batch + Group)
The following MA plot shows how most of the housekeeping genes, circled in orange, are not lying near the X axis, although the results for them show that they are not significantly differentially expressed (except 1 out of the 40).
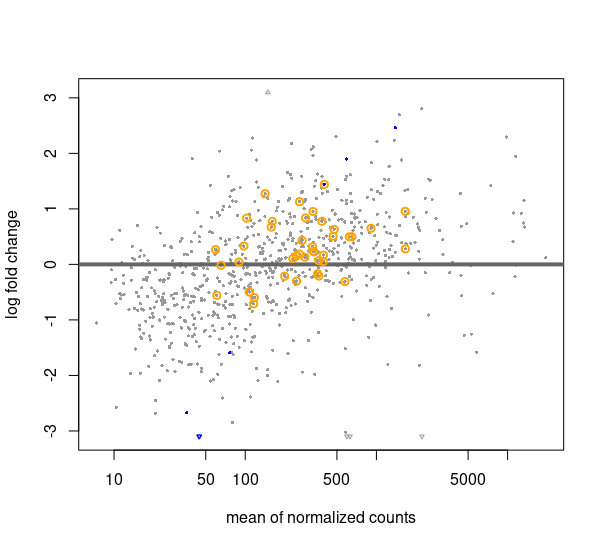
My questions are:
1) Should I be using these "housekeeping genes" as reference or I cannot trust them because of the previous check? 2) If the answer to 1 is "no", what alternatives do I have? What is the next step towards differential expression in this dataset? Maybe another way of defining housekeeping genes? 3) Is the use of the known "Batch" variable correct in this case? Or is it redundant with the previously estimated RUVg factors?
Any help is much appreciated. Thanks!

Hello Michael, and thank you for your reply.
I have done as you described, estimating size factors with controlGenes before DESeq, but the resulting MA plot is not much different from before. For the plot I attach I am still using the RUVg factors in the design formula. Am I supposed to discard those factors since I am already normalizing using housekeeping genes as reference?
This looks as expected with the control genes centered on the x-axis (yes, these can have high variance, this is discussed in work by Risso and Dudoit for example).
This part of the process is separate from inclusion of RUVg factors in the design -- you should do both.