
Hi all,
I understand WGCNA created to assess gene expression data, however; I have noticed that this method has been applied to microbial communities in some studies (Duran-Pinedo et al., 2011; Aylward et al., 2015; Guidi et al., 2016; Wilson et al., 2018).
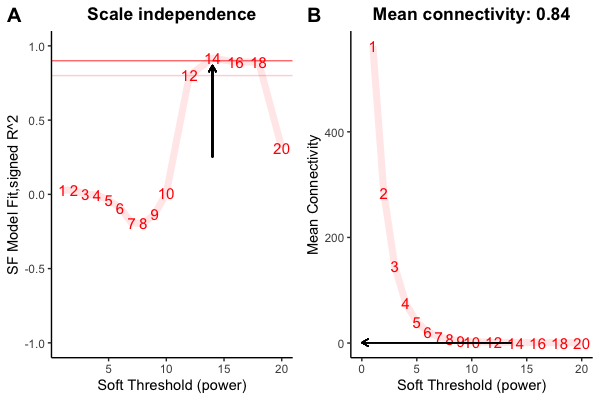
I am trying to do WGCNA for microbial data (ASV abundance matrix). After doing filtering and removed the low abundant taxa (ASV), I have done the analsyis and picked the soft threshold at 14. As you can see in the figure below-
and then -
modules.Y <- blockwiseModules(omics_data,
power = 14,
networkType = "signed",
TOMType = "signed",
corType = "bicor",
maxPOutliers = 0.05,
deepSplit = 4, # Default 2
minModuleSize = 10, # Default 30
minCoreKME = 0.5, # Default 0.5
minCoreKMESize = 2, # Default minModuleSize/3,
minKMEtoStay = 0.5, # Default 0.3
reassignThreshold = 0, # Default 1e-6
mergeCutHeight = 0.2, # Default 0.15
pamStage = FALSE,
pamRespectsDendro = TRUE,
replaceMissingAdjacencies = TRUE,
numericLabels = TRUE,
saveTOMFileBase = "TOM",
verbose = 3,
nThreads = 10,
maxBlockSize=8000)
I observed 24 modules. So far good (I suppose) 😊
Because my data has batch effect, therefore I removed the batch effect from my data and done the same analysis as mentioned above and observe that my soft threshold is going beyond 30 -
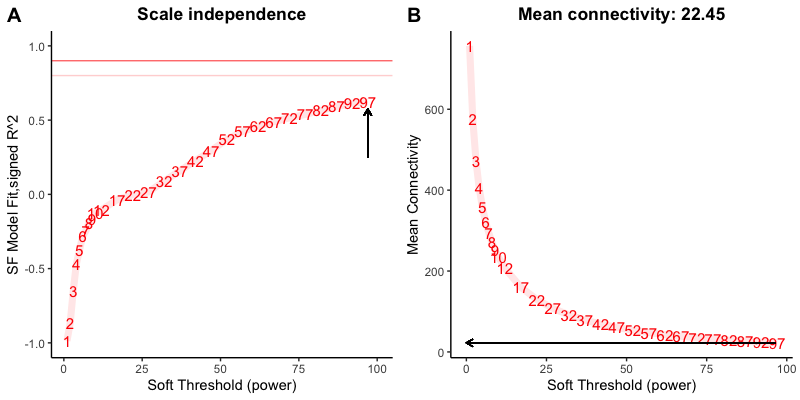
For downstream analysis, I can not provide the power over 30, therefore I used 30 as a soft threshold to generate 11 modules.
So, what would you recommend in this case? Do you think it's okay to use 30 (soft threshold)?