
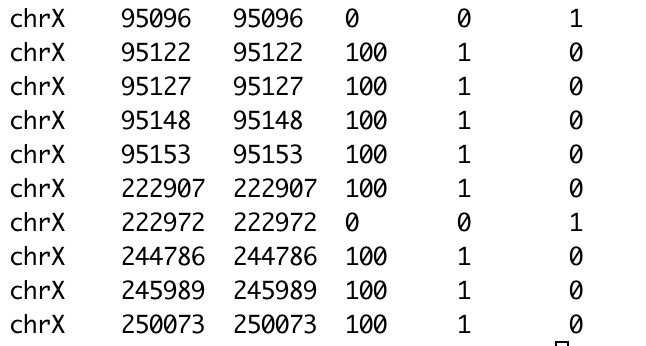
Hi, I use the methread function to read the coverage files from Bismark. BS_21=methRead("2368_bismark_bt2_pe.deduplicated.bismark.cov.gz", sample.id="2368_BS",assembly="mm10", header=F,skip=0,sep="\t",context="CpG", pipeline="bismarkCoverage", mincov = 5) However, the resulting methylkit object seems to lose the methylation percentage values generated by Bismark.
{kind=link}
MethyKitObject: https://user-images.githubusercontent.com/64626735/161638488-5652f1a1-92ee-4716-87c6-f103e093c960.png
{kind=link}
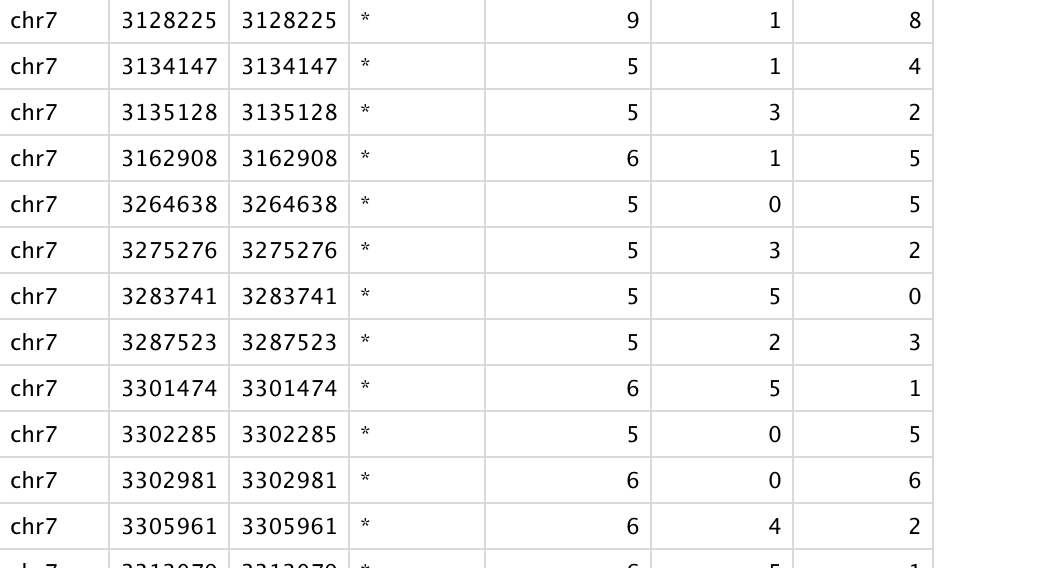
While I have found out a quick way of incorporating the meth %s for each methylation call from BS-seq experiments; the same is tricky when it comes to 5hmC data that I derive using the adjustmethylC function (because then I have two meth %s; one from BS and other from OXBS).
Would a plain subtraction of percent.meth per CpG (derived from Bismark) (BS minus OXBS) be the ideal way to go?
N.B. As expected, I have way more methylation calls from BS than the OXBS experiment.
I think that this whole situation stems from the fact that after running methread; the percentage methylation values of the coverage output from Bismark are lost.
My end goal is to generate 5mC and 5hmc signals per gene.
Please advise.