
Hi
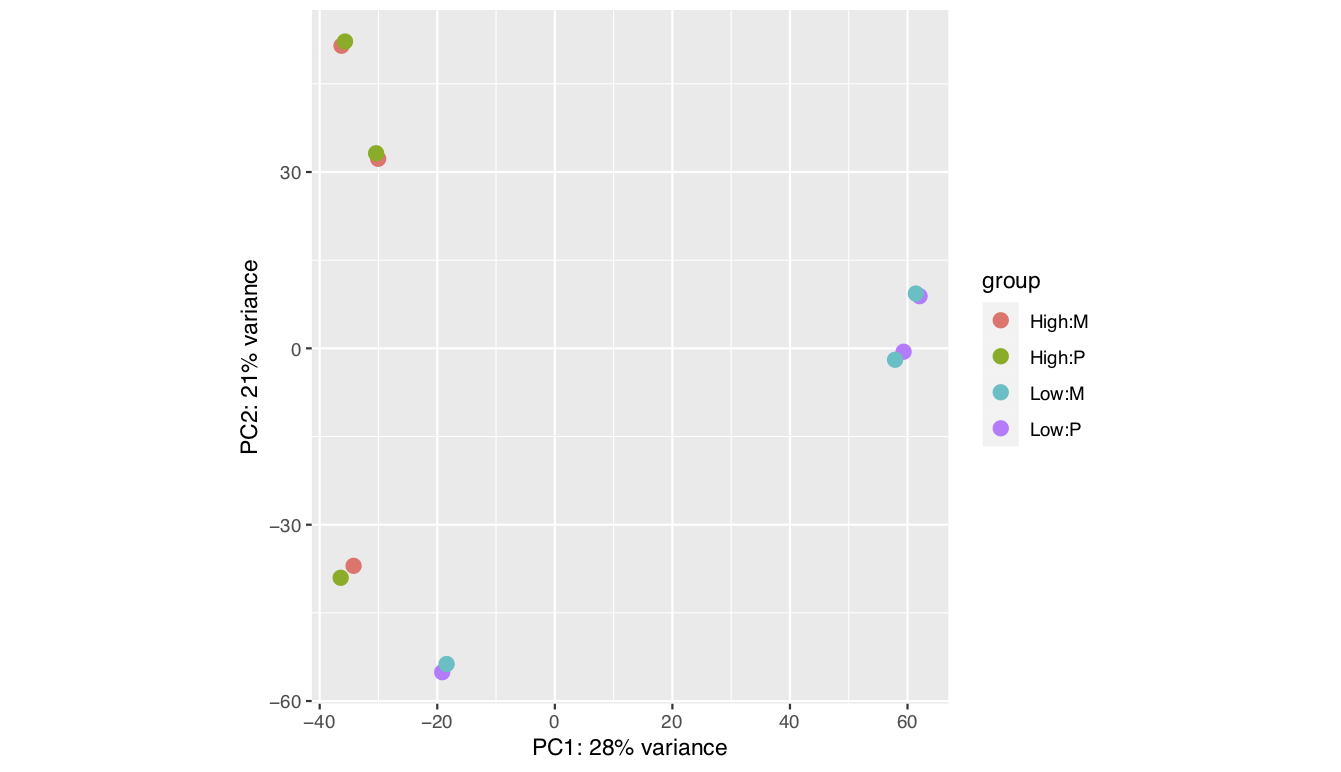
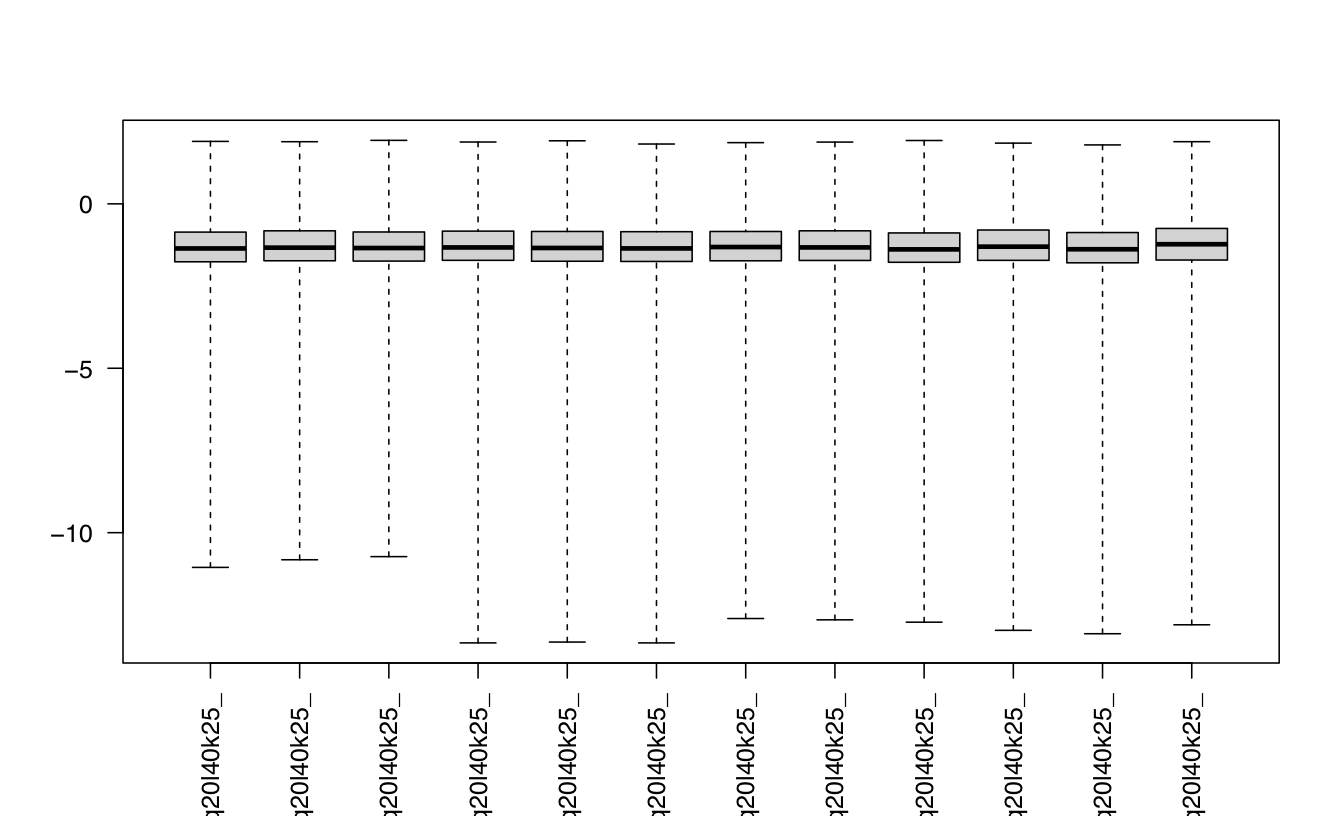
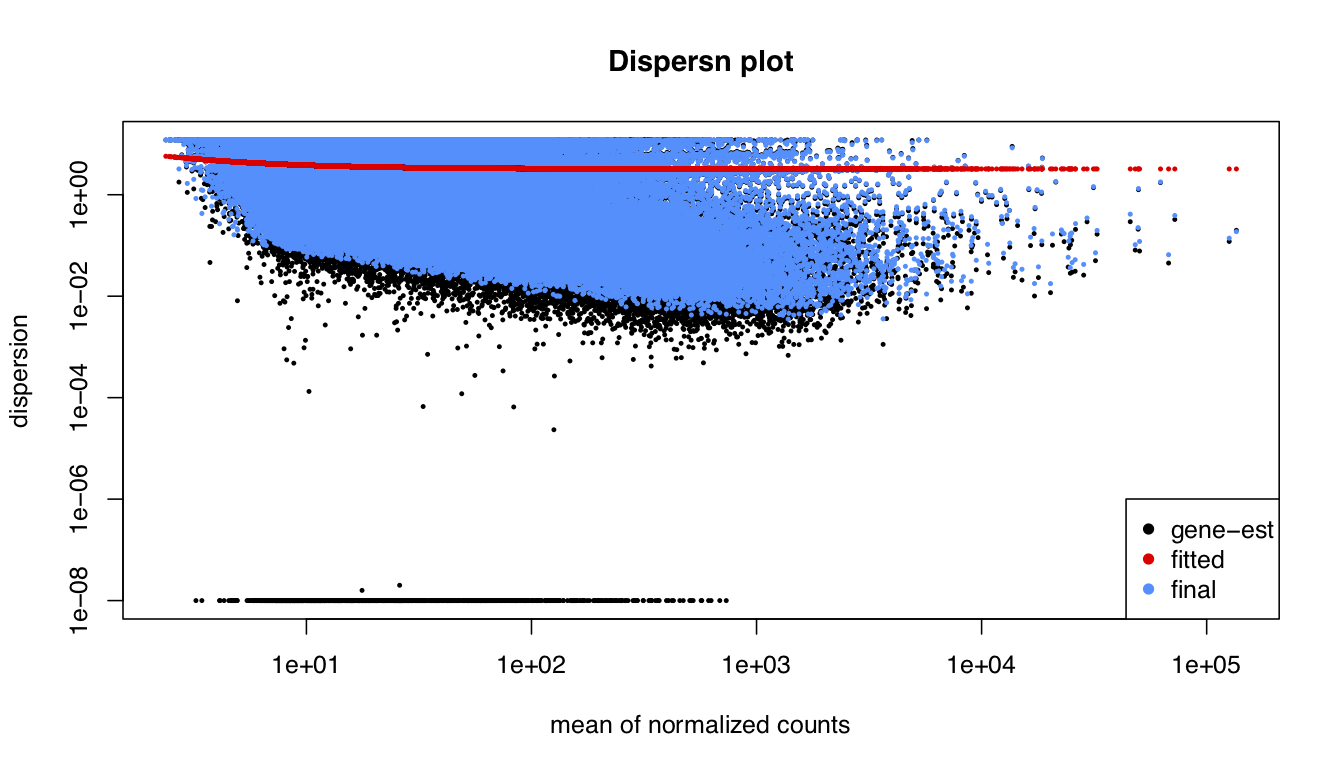
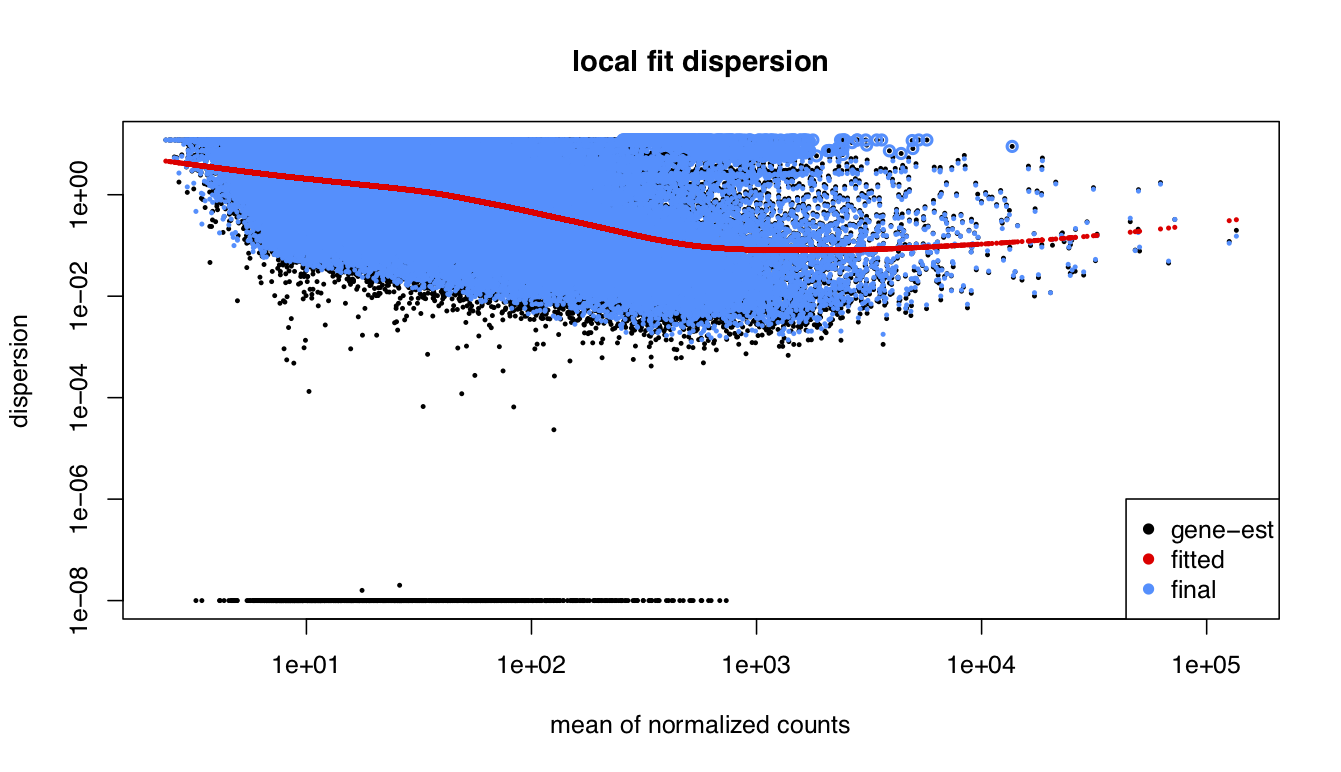
I been analysing some bulk rnaseq data of a non-model plant, looking at two treatments and two tissue stages. We used transcripts and salmon counts for looking at DEGs. Looking through the pca, cooks and dispersion plot (see below) – I been wondering if there is a concern of potential outlier samples/high variance as I notice one sample from each stage is away from the other two samples. The biological replicates are from three different trees – so there will be an element of biological variance due to that. So to account for that I have tried to fit two surrogate variables using sva (Note: I have also filtered some of the low count samples). Another concern was the dispersion plot – noticed the fitted line was one but this improves when a ‘local fit’ has been carried out. So I been wondering if it’s safe to go ahead and analyse the DEGs as is and potential bias I may be carrying forward. So was wondering if anyone could advice on this. Any thoughts would be much appreciated. Thanks in advance
#Create deseq object and run deseq
ddsTxi <- DESeqDataSetFromTximport(txi,colData = samples, design = ~ trt + Tissue)
ddsTxi
dds <- DESeq(ddsTxi)
#check data with plots
vsd <- vst(dds,blind=FALSE)
plotPCA(vsd,intgroup=c("trt","Tissue"))
#look at cooks distances
cooks <- log10(assays(dds)[["cooks"]])
boxplot(cooks, range=0, las=2)
nrow(dds)
keep <- rowSums(counts(dds) >= 10) >= 3 #at least3 samples with a count of 10 or higher
dds_filt <- dds[keep,]
dds_filt_loc <- estimateDispersions(dds_filt, fitType = "local")
nrow(dds_filt)
#percentage removed
nrow(dds_filt)/ nrow(dds)
vsd <- vst(dds_filt,blind=FALSE)
plotPCA(vsd,intgroup=c("trt","Tissue"))
#Disperssion plot on collapsed data
plotDispEsts(dds_filt, main="Dispersn plot")
plotDispEsts(dds_filt_loc,main="local fit dispersion")
#SVA
dat <- counts(dds_filt, normalized = TRUE)
idx <- rowMeans(dat) > 1
dat <- dat[idx, ]
mod <- model.matrix(~ trt + Tissue, colData(dds_filt))
mod0 <- model.matrix(~ 1, colData(dds_filt))
svseq <- svaseq(dat, mod, mod0, n.sv = 2)
svseq$sv
par(mfrow = c(2, 1), mar = c(3,5,3,1))
for (i in 1:2) {
stripchart(svseq$sv[, i] ~ dds_filt$trt, vertical = TRUE, main = paste0("SV", i))
abline(h = 0)
}

Thanks for the reply, no worries.
There is a possibility the trees are driving some of the grouping (though ideally we would like to see two groupings not three) - the plants are of wild population - non clonal. Though they are of same species, we are suspecting they may be of different genotypes. Hence trying to remove some the unwanted variation using
sva. But through that I didn't see a major improvement (see stripchart below). Any suggestions would be much appreciatedDid you try refitting the model while controlling for the SVs? Eg put the two SVs in the design, and test on your variables of interest. This should help you focus your tests on the explainable part of the count variation.
Yep, so far that's how I have carried forward with the analysis - I also used a local fit based on the dispersion plot. However I haven't seen any improvement with the groupings seen in PCA plot. Is there anything more I could do?
Haven't seen improvement in PCA plot -- see the FAQ in vignette on this topic.