
Dear DESeq2 experts,
I hope my message finds you fine. I am facing a problem in carrying out differential gene expression analysis using DESeq2 in a scRNA-seq dataset. I have clustered cells into distinct clusters and created pseudobulk counts by summing gene read counts of cells in a given cluster in each sample and would like to carry out differential expression analysis using these pseudobulks.
The problem I am facing now is that the cell counts as well as the total read counts (after count aggregation) across the different samples greatly vary and are very much unbalanced as you can see in the attached graphs.
Now, if I run DEseq2, I get no significantly differentially expressed genes between the two comparsion groups I am interested in.
Is there a solution to this problem? Can one still use DESeq2 to analyze differential expression in such a scenario?
*The graphs here show the number of cells and the total number of reads across samples in one particular cell cluster. The red and green colors indicate the two different groups I would like to compare.
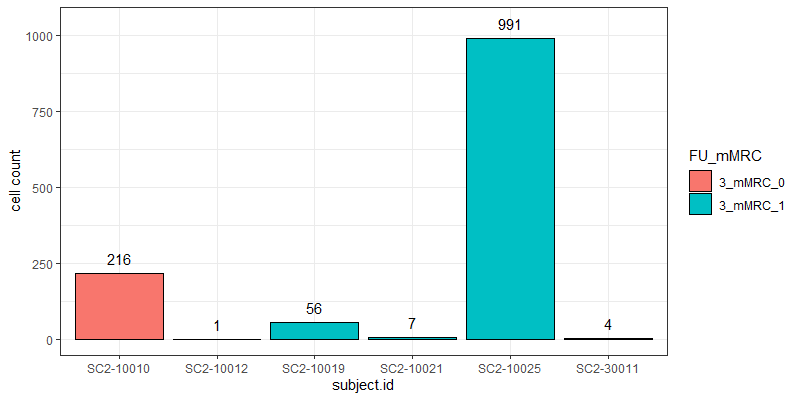
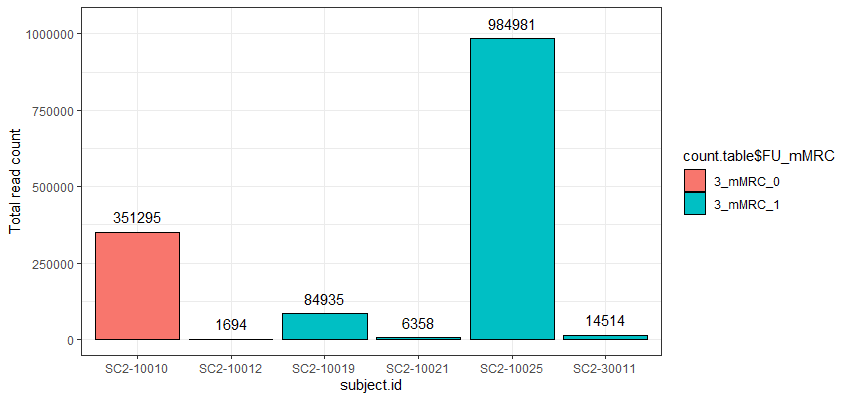

Thank you very much for your kind opinion. I think setting a threshold for the minimum cluster size to include in the analysis is a good way to deal with this problem :)