
I am trying out different DiffBind normalization methods for my ATAC-seq dataset in order to select the most suitable one. However, it looks like no matter which normalization method from the official manual I use, it always reverts to default normalization method.
To illustrate (starting with the default - normalization using full library sizes, DESeq2 method):
> normbyRD <- dba.normalize(non_norm_data, normalize=DBA_NORM_LIB)
> normbyRD
6 Samples, 54620 sites in matrix:
ID Tissue Treatment Replicate Reads FRiP
1 1a Muscle WT 1 19852956 0.35
2 2a Muscle WT 2 32243037 0.26
3 3a Muscle WT 3 36275376 0.29
4 1ab Muscle Treated 1 37432844 0.26
5 2ab Muscle Treated 2 40729414 0.28
6 3ab Muscle Treated 3 37136858 0.27
Design: [~Treatment] | 1 Contrast:
Factor Group Samples Group2 Samples2
1 Treatment Treated 3 WT 3
> dba.normalize(normbyRD, method = DBA_DESEQ2, bRetrieve = TRUE)
$norm.method
[1] "lib"
$norm.factors
[1] 0.5848552 0.9498589 1.0686490 1.1027472 1.1998620 1.0940277
$lib.method
[1] "full"
$lib.sizes
[1] 19852956 32243037 36275376 37432844 40729414 37136858
$control.subtract
[1] TRUE
$filter.value
[1] 1
> normbyRD <- dba.analyze(normbyRD)
Applying Blacklist/Greylists...
Genome detected: Hsapiens.UCSC.hg38
Applying blacklist...
Removed: 136 of 54620 intervals.
Removed 136 (of 54620) consensus peaks.
Normalize DESeq2 with defaults...
Analyzing...
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
When I try to normalize with both edgeR and DESeq2 approaches but using Reads in Peaks
> normbyRIP <- dba.normalize(non_norm_data, method=DBA_ALL_METHODS, normalize=DBA_NORM_NATIVE, library=DBA_LIBSIZE_PEAKREADS)
> normbyRIP
...[output here same as above for > normbyRD]
> dba.normalize(normbyRIP, method=DBA_DESEQ2, bRetrieve=TRUE)
$norm.method
[1] "RLE"
$norm.factors
1a 2a 3a 1ab 2ab 3ab
0.6154949 0.8637777 1.0765341 1.1687352 1.3005265 1.1629528
$lib.method
[1] "RiP"
$lib.sizes
1a 2a 3a 1ab 2ab 3ab
6918883 8231661 10701169 9759950 11282510 9910829
$control.subtract
[1] TRUE
$filter.value
[1] 1
> dba.normalize(normbyRIP, method=DBA_EDGER, bRetrieve=TRUE)
$norm.method
[1] "TMM"
$norm.factors
[1] 0.8358383 0.9751437 0.9405268 1.1106895 1.0755978 1.0919314
$lib.method
[1] "RiP"
$lib.sizes
1a 2a 3a 1ab 2ab 3ab
6918883 8231661 10701169 9759950 11282510 9910829
$control.subtract
[1] TRUE
$filter.value
[1] 1
So far everything looks like it's working.
But after I run the following, peaks and their numbers from "DESeq2 full library" method are the same as from "DESeq2 RiP" method
> normbyRIP <- dba.analyze(normbyRIP, method=DBA_ALL_METHODS)
Applying Blacklist/Greylists...
Genome detected: Hsapiens.UCSC.hg38
Applying blacklist...
Removed: 136 of 54620 intervals.
Removed 136 (of 54620) consensus peaks.
Normalize edgeR with defaults...
Normalize DESeq2 with defaults...
Analyzing...
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
Here is the Venn diagram (MA plots also look identical)
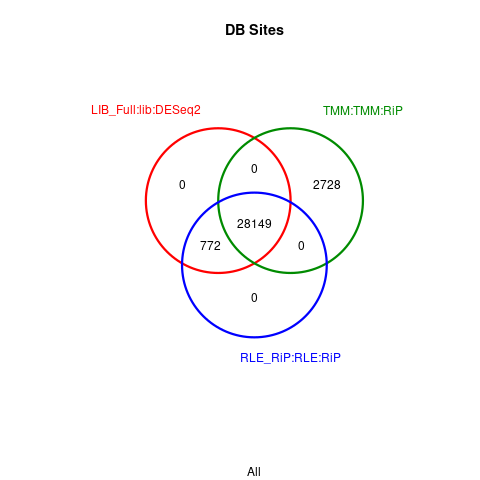
Same happens when I try to use other normalization methods (background etc.)
I do not know what is causing this but these lines suggest that dba.analyze somehow overwrites the method that I chose for normalization.
Normalize edgeR with defaults...
Normalize DESeq2 with defaults...
Would you be able to explain that, please, Rory Stark?
> sessionInfo()
R version 4.1.3 (2022-03-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=en_GB.UTF-8 LC_NUMERIC=C LC_TIME=en_GB.UTF-8 LC_COLLATE=en_GB.UTF-8 LC_MONETARY=en_GB.UTF-8
[6] LC_MESSAGES=en_GB.UTF-8 LC_PAPER=en_GB.UTF-8 LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] ChIPpeakAnno_3.28.1 edgeR_3.36.0 limma_3.50.3 BiocParallel_1.28.3 DiffBind_3.4.11
[6] SummarizedExperiment_1.24.0 Biobase_2.54.0 MatrixGenerics_1.6.0 matrixStats_0.62.0 GenomicRanges_1.46.1
[11] GenomeInfoDb_1.30.1 IRanges_2.28.0 S4Vectors_0.32.4 BiocGenerics_0.40.0
loaded via a namespace (and not attached):
[1] amap_0.8-18 colorspace_2.0-3 rjson_0.2.21 hwriter_1.3.2.1 ellipsis_0.3.2
[6] futile.logger_1.4.3 XVector_0.34.0 ggrepel_0.9.1 bit64_4.0.5 AnnotationDbi_1.56.2
[11] fansi_1.0.3 mvtnorm_1.1-3 apeglm_1.16.0 xml2_1.3.3 splines_4.1.3
[16] cachem_1.0.6 geneplotter_1.72.0 Rsamtools_2.10.0 annotate_1.72.0 dbplyr_2.1.1
[21] ashr_2.2-54 png_0.1-7 graph_1.72.0 GreyListChIP_1.26.0 compiler_4.1.3
[26] httr_1.4.2 lazyeval_0.2.2 assertthat_0.2.1 Matrix_1.4-1 fastmap_1.1.0
[31] cli_3.2.0 formatR_1.12 prettyunits_1.1.1 htmltools_0.5.2 tools_4.1.3
[36] coda_0.19-4 gtable_0.3.0 glue_1.6.2 GenomeInfoDbData_1.2.7 systemPipeR_2.0.8
[41] dplyr_1.0.8 rappdirs_0.3.3 ShortRead_1.52.0 Rcpp_1.0.8.3 bbmle_1.0.24
[46] vctrs_0.4.1 Biostrings_2.62.0 multtest_2.50.0 rtracklayer_1.54.0 stringr_1.4.0
[51] lifecycle_1.0.1 irlba_2.3.5 ensembldb_2.18.4 restfulr_0.0.13 gtools_3.9.2
[56] InteractionSet_1.22.0 XML_3.99-0.9 zlibbioc_1.40.0 MASS_7.3-56 scales_1.2.0
[61] BSgenome_1.62.0 ProtGenerics_1.26.0 hms_1.1.1 RBGL_1.70.0 parallel_4.1.3
[66] AnnotationFilter_1.18.0 lambda.r_1.2.4 RColorBrewer_1.1-3 curl_4.3.2 yaml_2.3.5
[71] memoise_2.0.1 ggplot2_3.3.5 emdbook_1.3.12 biomaRt_2.50.3 bdsmatrix_1.3-4
[76] latticeExtra_0.6-29 stringi_1.7.6 RSQLite_2.2.12 SQUAREM_2021.1 genefilter_1.76.0
[81] BiocIO_1.4.0 filelock_1.0.2 GenomicFeatures_1.46.5 caTools_1.18.2 truncnorm_1.0-8
[86] rlang_1.0.2 pkgconfig_2.0.3 bitops_1.0-7 lattice_0.20-45 invgamma_1.1
[91] purrr_0.3.4 GenomicAlignments_1.30.0 htmlwidgets_1.5.4 bit_4.0.4 tidyselect_1.1.2
[96] plyr_1.8.7 magrittr_2.0.3 DESeq2_1.34.0 R6_2.5.1 gplots_3.1.1
[101] generics_0.1.2 DelayedArray_0.20.0 DBI_1.1.2 pillar_1.7.0 survival_3.3-1
[106] KEGGREST_1.34.0 RCurl_1.98-1.6 mixsqp_0.3-43 tibble_3.1.6 crayon_1.5.1
[111] futile.options_1.0.1 KernSmooth_2.23-20 utf8_1.2.2 BiocFileCache_2.2.1 jpeg_0.1-9
[116] progress_1.2.2 locfit_1.5-9.5 grid_4.1.3 blob_1.2.3 digest_0.6.29
[121] VennDiagram_1.7.3 xtable_1.8-4 regioneR_1.26.1 numDeriv_2016.8-1.1 munsell_0.5.0

I've checked in a fix for this (
DiffBind_3.6.1). DiffBind will now re-normalize after applying blacklists/greylists, using the originally specified parameters.Thanks a lot, everything works now!