Entering edit mode

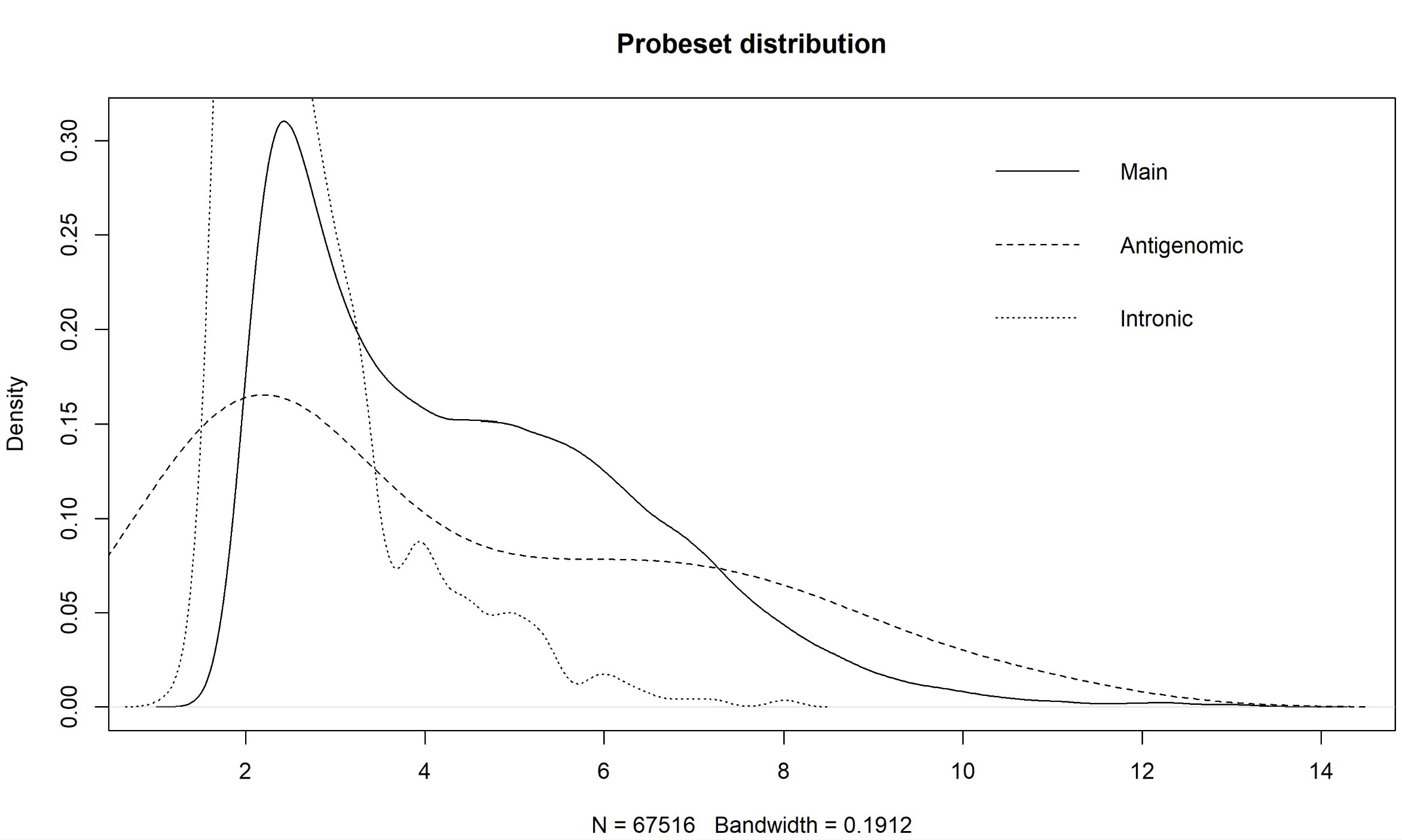
Dear Bioconductor members, I'm asked to re-analyze the microarray data of GSE76297 (CEL). As far as I know, paCalls can not be used in HTAFeatureSet. And there is a method based on density plot of main, intronic, and antigenomic probesets by Prof. James W. MacDonald (Appropriate pre-processing pipeline for Human Transcriptome Array HTA 2.0 with oligo for DE analysis). However, there are still some questions about this methods. Why is the cutoff based on intronic probesets? What is the purpose for the antigenomic probesets here for filtering? And are there quantitative methods like paCalls to filtering the unexpressed probes? Thanks in advance! Yang Shi
eset <- oligo::rma(object = affyRaw, target = 'core')
eset.main <- getMainProbes(input = eset, level = 'core')
require(hta20transcriptcluster.db)
eset.main <- annotateEset(eset.main, hta20transcriptcluster.db)
plot(density(exprs(eset.main)), main = "Probeset distribution", xaxt = 'n')
for(i in c(2,7)) lines(density(exprs(eset.main)[as.character(probeType[probeType[,2] %in% i,1]),1]), lty = if(i == 2) 2 else 3)
legend("topright", c("Main","Antigenomic","Intronic"), lty = 1:3, bty="n")
axis(1, xaxp=c(1,14,15), las=2)
R version 4.2.0 (2022-04-22 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19043)
Matrix products: default
locale:
[1] LC_COLLATE=Chinese (Simplified)_China.utf8
[2] LC_CTYPE=Chinese (Simplified)_China.utf8
[3] LC_MONETARY=Chinese (Simplified)_China.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=Chinese (Simplified)_China.utf8
attached base packages:
[1] stats4 stats graphics utils datasets grDevices
[7] methods base

Perfect answer! This questions troubled me for a long time. Thanks again and for your quick and detailed reply.