Entering edit mode

Dear Bioconductor members, CEL files of Affymetrix could be normalized after oligo::rma. Is it necesssary to perform quantile normalization by limma::normalizeBetweenArrays? Here is the boxplot showing every step and the related clustering results (red = normal samples, black = tumor samples). Thanks in advance!
eset <- oligo::rma(object = affyRaw, target = 'core')
require(affycoretools)
eset <- getMainProbes(input = eset, level = 'core')
boxplot(exprSet, outline = FALSE, las = 2)
dist_mat = dist(x = t(exprs(eset)), method = 'euclidean')
res.hc = hclust(d = dist_mat, method = 'complete')
labels = res.hc[['labels']]
res.hc[['labels']] = as.character(metadata[labels,meta])
label_color = as.numeric(as.factor(labels(res.hc)))
fviz_dend(x = res.hc,label_cols = label_color,cex = 0.6)
exprSet <- limma::normalizeBetweenArrays(object = exprSet)
boxplot(exprSet, outline = FALSE, las = 2)
dist_mat = dist(x = t(exprs(eset)), method = 'euclidean')
res.hc = hclust(d = dist_mat, method = 'complete')
labels = res.hc[['labels']]
res.hc[['labels']] = as.character(metadata[labels,meta])
label_color = as.numeric(as.factor(labels(res.hc)))
fviz_dend(x = res.hc,label_cols = label_color,cex = 0.6)
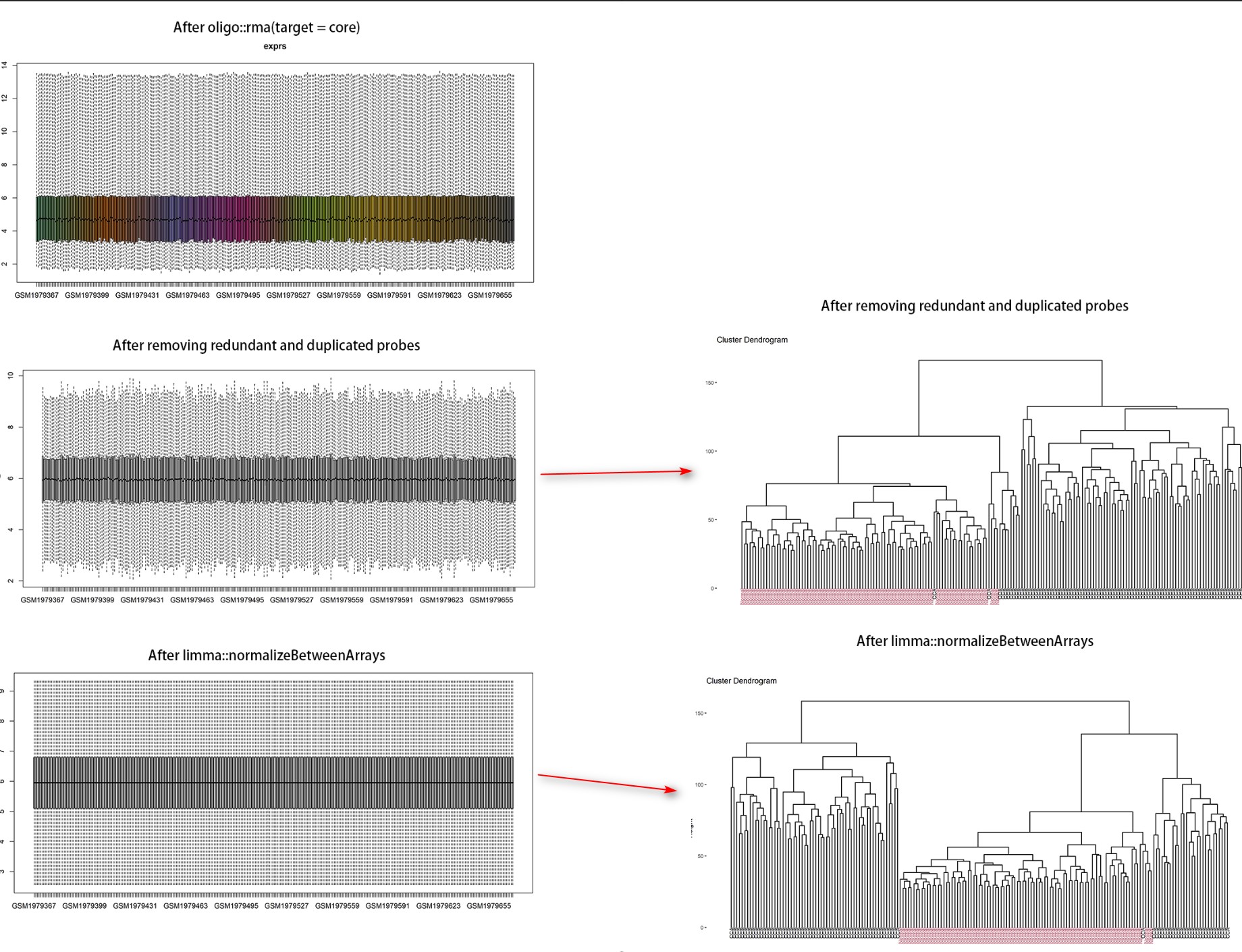

Thanks you so much sir! But I'm still confused with the boxplot showing here. Why oligo::rma could not get the plot like limma::normalizeBetweenArrays? In my opinion, the normalization was judged by the median line at the same level in the boxplot.
Affymetrix arrays have multiple probes that are meant to measure each transcript and are summarized to give an overall estimate of the transcript abundance. The RMA algorithm first background corrects the probe intensities, then normalizes the individual probes using a quantile normalization, and then summarizes using Tukey's median polish algorithm. Since the normalization takes place before the summarization step you don't get the same distribution of data that you get if you use a quantile normalization on the summarized values.
I will also point out that this is sort of a settled point. In the early 2000's many people proposed different methods to process Affymetrix arrays, and in the end the RMA algorithm became the de facto standard. Since then, if you analyze some Affy data and then in your manuscript you say 'we used the RMA algorithm' and point to the paper that first described the process, the reviewers will sort of nod sagely and move on. If instead you say 'we used the RMA algorithm and then followed up with an additional quantile normalization' then an astute reviewer will have questions about that, and will likely also wonder if you have done other things that need extra attention.
Got that sir! Really thank you!