There are some QC probes that you might use.
> library(pd.hg.u219)
> library(DBI)
> con <- db(pd.hg.u219)
> dbListTables(con)
[1] "featureSet" "pmfeature" "table_info"
> dbGetQuery(con, "select * from featureSet where man_fsetid LIKE 'AFFX%';")
fsetid strand man_fsetid
1 1 1 AFFX-DapX-5_at
2 2 1 AFFX-DapX-M_at
3 3 1 AFFX-DapX-3_at
4 4 1 AFFX-LysX-5_at
5 5 1 AFFX-LysX-M_at
6 6 1 AFFX-LysX-3_at
7 7 1 AFFX-PheX-5_at
8 8 1 AFFX-PheX-M_at
9 9 1 AFFX-PheX-3_at
10 10 1 AFFX-ThrX-5_at
11 11 1 AFFX-ThrX-M_at
12 12 1 AFFX-ThrX-3_at
13 13 1 AFFX-TrpnX-5_at
14 14 1 AFFX-TrpnX-M_at
15 15 1 AFFX-TrpnX-3_at
16 16 1 AFFX-r2-Ec-bioB-5_at
17 17 1 AFFX-r2-Ec-bioB-M_at
18 18 1 AFFX-r2-Ec-bioB-3_at
19 19 1 AFFX-r2-Ec-bioC-5_at
20 20 1 AFFX-r2-Ec-bioC-3_at
21 21 1 AFFX-r2-Ec-bioD-5_at
22 22 1 AFFX-r2-Ec-bioD-3_at
23 23 1 AFFX-r2-P1-cre-5_at
24 24 1 AFFX-r2-P1-cre-3_at
25 25 1 AFFX-r2-Bs-dap-5_at
26 26 1 AFFX-r2-Bs-dap-M_at
27 27 1 AFFX-r2-Bs-dap-3_at
28 28 1 AFFX-r2-Bs-lys-5_at
29 29 1 AFFX-r2-Bs-lys-M_at
30 30 1 AFFX-r2-Bs-lys-3_at
31 31 1 AFFX-r2-Bs-phe-5_at
32 32 1 AFFX-r2-Bs-phe-M_at
33 33 1 AFFX-r2-Bs-phe-3_at
34 34 1 AFFX-r2-Bs-thr-5_s_at
35 35 1 AFFX-r2-Bs-thr-M_s_at
36 36 1 AFFX-r2-Bs-thr-3_s_at
37 37 1 AFFX-HUMISGF3A/M97935_5_at
38 38 1 AFFX-HUMISGF3A/M97935_MA_at
39 39 1 AFFX-HUMISGF3A/M97935_MB_at
40 40 1 AFFX-HUMISGF3A/M97935_3_at
41 41 1 AFFX-HUMRGE/M10098_5_at
42 42 1 AFFX-HUMRGE/M10098_M_at
43 43 1 AFFX-HUMRGE/M10098_3_at
44 44 1 AFFX-HUMGAPDH/M33197_5_at
45 45 1 AFFX-HUMGAPDH/M33197_M_at
46 46 1 AFFX-HUMGAPDH/M33197_3_at
47 47 1 AFFX-HSAC07/X00351_5_at
48 48 1 AFFX-HSAC07/X00351_M_at
49 49 1 AFFX-HSAC07/X00351_3_at
50 50 1 AFFX-M27830_5_at
51 51 1 AFFX-M27830_M_at
52 52 1 AFFX-M27830_3_at
53 53 1 AFFX-hum_alu_at
54 54 1 AFFX-r2-TagA_at
55 55 1 AFFX-r2-TagB_at
56 56 1 AFFX-r2-TagC_at
57 57 1 AFFX-r2-TagD_at
58 58 1 AFFX-r2-TagE_at
59 59 1 AFFX-r2-TagF_at
60 60 1 AFFX-r2-TagG_at
61 61 1 AFFX-r2-TagH_at
62 62 1 AFFX-r2-TagJ-3_at
63 63 1 AFFX-r2-TagJ-5_at
64 64 1 AFFX-r2-TagO-3_at
65 65 1 AFFX-r2-TagO-5_at
66 66 1 AFFX-r2-TagQ-3_at
67 67 1 AFFX-r2-TagQ-5_at
68 68 1 AFFX-r2-TagIN-3_at
69 69 1 AFFX-r2-TagIN-M_at
70 70 1 AFFX-r2-TagIN-5_at
71 71 1 AFFX-Nonspecific-GC03_at
72 72 1 AFFX-Nonspecific-GC04_at
73 73 1 AFFX-Nonspecific-GC05_at
74 74 1 AFFX-Nonspecific-GC06_at
75 75 1 AFFX-Nonspecific-GC07_at
76 76 1 AFFX-Nonspecific-GC08_at
77 77 1 AFFX-Nonspecific-GC09_at
78 78 1 AFFX-Nonspecific-GC10_at
79 79 1 AFFX-Nonspecific-GC11_at
80 80 1 AFFX-Nonspecific-GC12_at
81 81 1 AFFX-Nonspecific-GC13_at
82 82 1 AFFX-Nonspecific-GC14_at
83 83 1 AFFX-Nonspecific-GC15_at
84 84 1 AFFX-Nonspecific-GC16_at
85 85 1 AFFX-Nonspecific-GC17_at
86 86 1 AFFX-Nonspecific-GC18_at
87 87 1 AFFX-Nonspecific-GC19_at
88 88 1 AFFX-Nonspecific-GC20_at
89 89 1 AFFX-Nonspecific-GC21_at
90 90 1 AFFX-Nonspecific-GC22_at
91 91 1 AFFX-Nonspecific-GC23_at
92 92 1 AFFX-Nonspecific-GC24_at
93 93 1 AFFX-Nonspecific-GC25_at
Those last ones with the Nonspecific in the name are probes that aren't meant to bind to anything but have increasing GC content. At a certain point the GC content gets high enough that the probes will bind to lots of things, so they aren't all useful. You can decide which ones you might want to use by doing something like
> boxplot(t(exprs(eset)[grep("Nonspecific", row.names(eset)),]), xaxt = "n")
> axis(1, at = 1:23, gsub("AFFX-Nonspecific-", "", grep("Nonspecific", row.names(eset), value = TRUE)), las = 2)
I would argue that up to maybe 13 or 14 is useful as a measure of background binding, and you could just remove any probes where a certain proportion of the probesets are below that level.



Thanks for your excellent reply sir! But there are still some points confused me. (1).dbGetQuery get 93 kinds of probes. Would be okay for just selected those "Nonspecific" for backgroud binding intensity? (2).As your suggested, 13 or 14 maybe used as background binding. What is the criteria? Just based on expression value, like less than 5? Here is the plot drawn by you code. Thanks so much sir.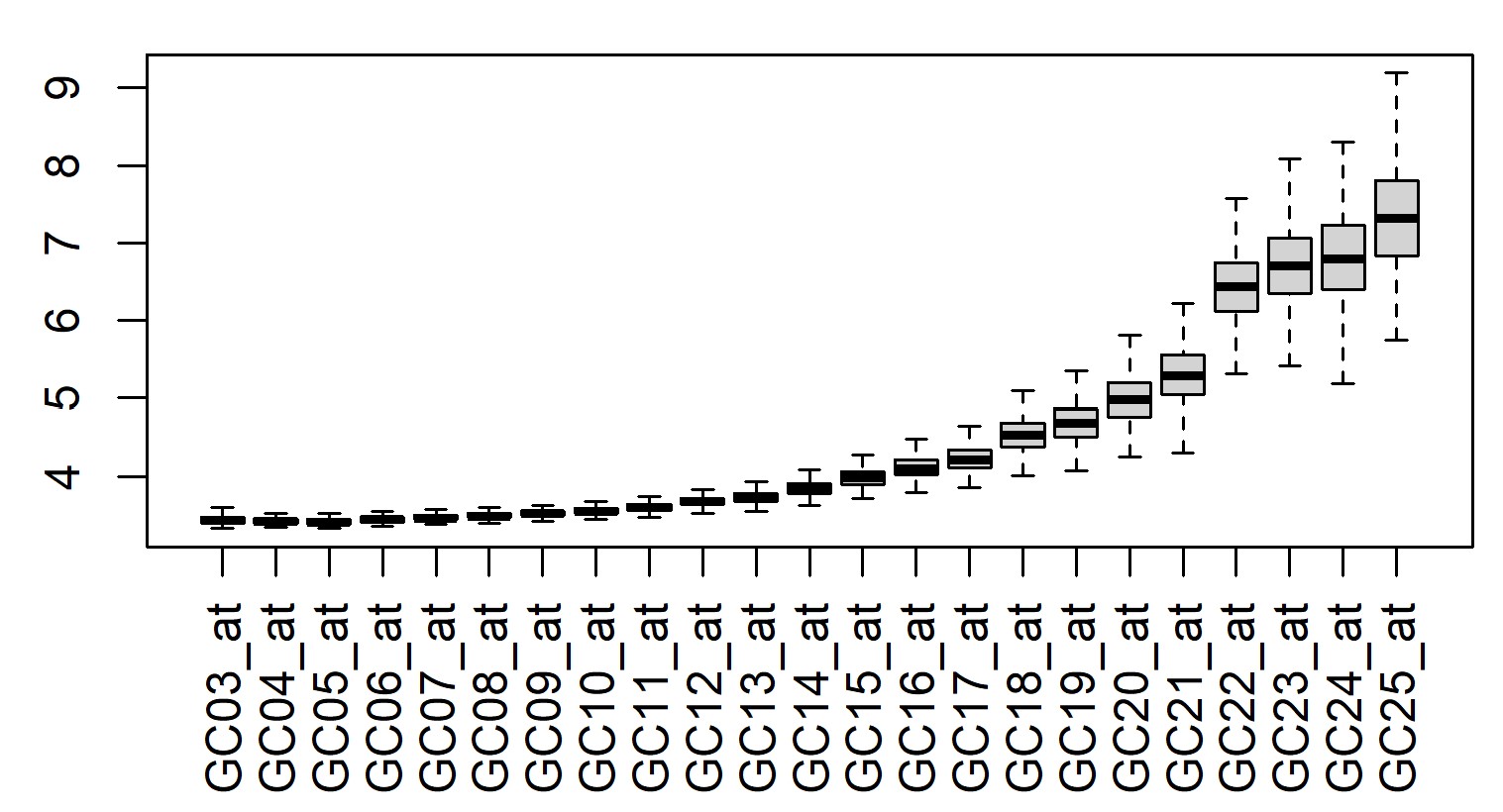
It's a non-scientific selection. The Nonspecific probes are meant to be sequences that are not found in nature, so any binding is due to cross-hybridization. This plot is one of the reasons that GCRMA was developed, as it's obvious that as the GC content goes up, the non-specific binding goes up as well.
But for your goal, you want to exclude probesets that you think are not really measuring anything, right? And these probes are not supposed to be measuring anything, but there is the added complexity that the high GC probes start to bind due to the GC issue. We need to define some expression level cutoff that represents background binding, without representing background binding that you expect when the GC content gets too high, so you have to decide at which point that starts to happen. I would argue 12 - 15, but it's your analysis, so you should decide for yourself.
Thanks sir! As your mentioned here, 12-15 means cutoff value about 4? Could I select the reflection point as the "point" that starts to happen?
We have progressed past the point where I show you how to do things using Bioconductor packages, and into the realm where I start telling you how to do your analysis, which is something I try really hard to avoid. As I mentioned in my last post, this is your analysis, and you want to exclude things, and I have simply showed you the probes that are arguably useful for doing that sort of thing.
What cutoffs you want to use are up to you to decide, because you are the one who will ultimately have to defend your choices.
Thanks sir! I got it now!