
Hey people, I’m analyzing scRNA-seq data for mice from 6 different biological groups. I am using Seurat (“MetaFeatures”/“AddModuleScore”) and UCell/ssGSEA (via “escape”) to try and look for differences in pathway/gene set representation between these groups. While looking at the results of hundreds of pathways/gene sets, I’ve noticed that most of these results look very similar to one another. I am now quite certain that – in most cases – the (many) differences I see between the experimental groups, in terms of their score for specific certain pathways/gene sets, are an artifact.
I suspect that the problem stems from differences (between the samples) in terms of the average number of unique genes (“nFeature”) and/or in terms of absolute cell numbers. I’m attaching an image with some graphs that exemplify the issue (I’ve removed group/set names, because I’m not allowed to reveal them). The top row includes the factors I suspect may cause the problem, while the bottom row includes UCell scores of a few gene sets that exemplify the problem (I’ve gotten similar results when using Seurat’s “MetaFeatures”/“AddModuleScore” functions). Also, as you can see, two of the six groups are from one batch (“Batch 1”) and the other 4 groups are from a different batch (“Batch 2”). Each group had its own (separate) lane on the 10X Chromium platform.
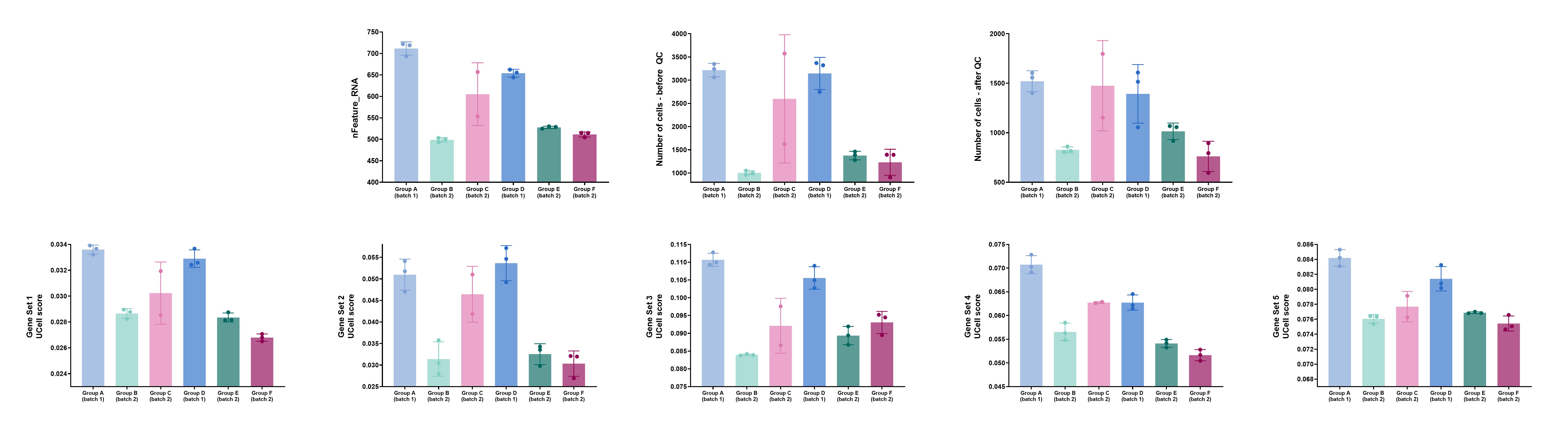
The data were normalized and integrated using Seurat before running the MetaFeatures/AddModuleScore/UCell/ssGSEA functions.
Any idea what I can do in order to remove these artifacts, so that I can get meaningful results?
Cheers,
Omer

To note: cross-posted at https://www.biostars.org/p/9554907/