Entering edit mode

I am trying to compare treatment (A) vs my control (C). A heavily downregulates genes, but for some reason my volcano plot shows that it is upregulated when compared to the control. The samples were not swapped (I've checked) and I got the correct distribution (downregulation) once, but since then have gotten the reverse. What is going wrong?
dds <- DESeqDataSetFromMatrix(countData = my_cts,
colData = my_coldata,
design = ~ condition)
dds
#prefilter row sums with less than 10 reads
keep <- rowSums(counts(dds)) >= 10
dds <- dds[keep,]
#factor levels
my_coldata$condition <- factor(my_coldata$condition, levels = c("C","A"))
#do i need these?
#dds <- estimateDispersions(dds)
#dds <- nbinomWaldTest(dds)
#run DESeq2
dds <- DESeq(dds)
#check coefficients estimated by DEseq
resultsNames(dds)
#obtain results
res <- results(dds, contrast=c("condition","A","C")) #order is treatment, numerator, denominator
#shrink LFC
resLFC <- lfcShrink(dds, coef = 2, type = "apeglm") #coef is set to "condition A vs C"
#make volcano plot (the result that looks flipped)
par(mfrow=c(1,1))
# Make a basic volcano plot
with(res, plot(log2FoldChange, -log10(pvalue), pch=20, main="Volcano plot", xlim=c(-4,4)))
with(subset(res, padj<.01 ), points(log2FoldChange, -log10(pvalue), pch=20, col="blue"))
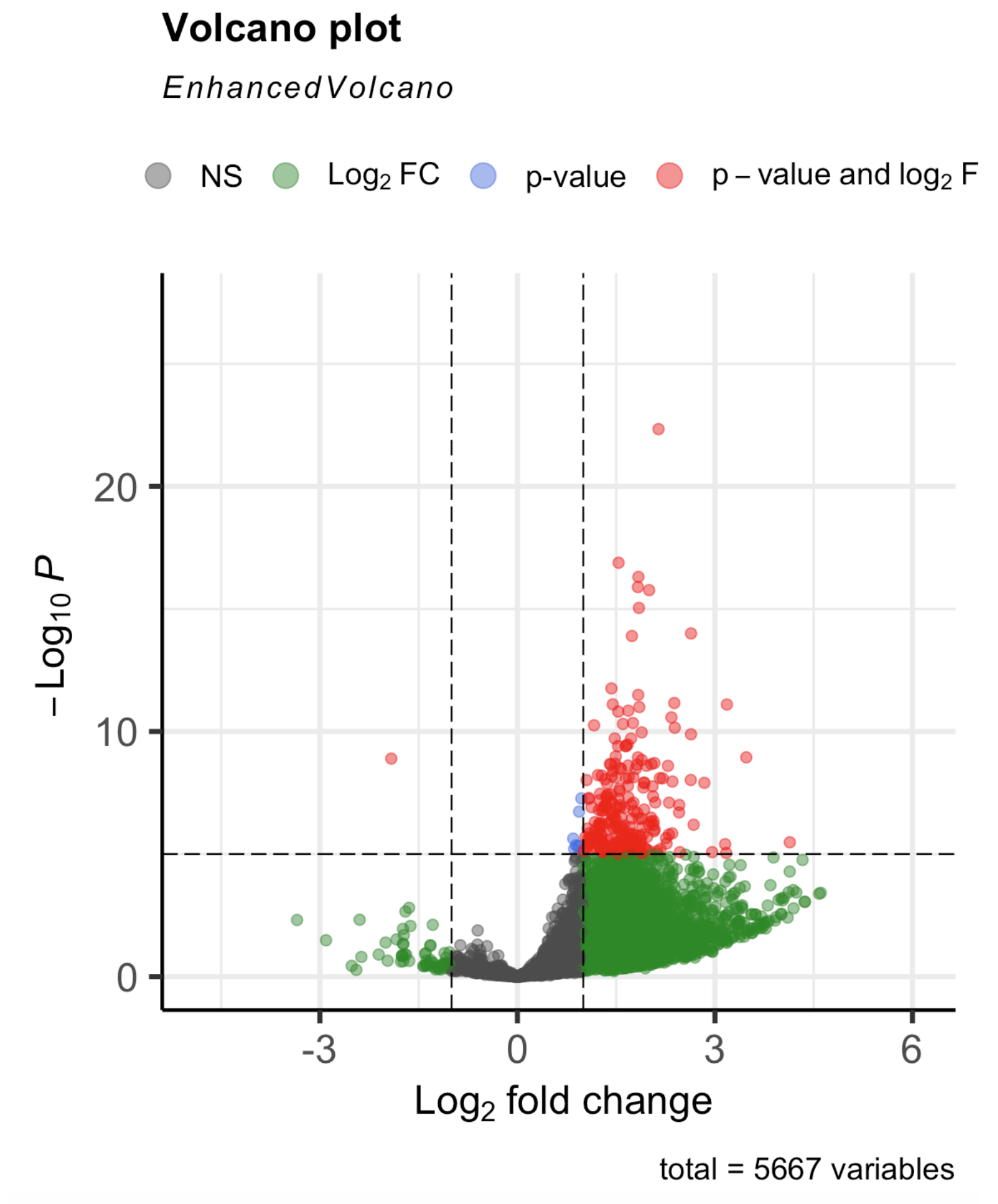
Any advice is much appreciated.

Hi Michael, so does this mean I have to factor dds$condition as well? I though I could get the same result by running:
res <- results(dds, contrast=c("condition","A","C"))If not, how do I modify the code so that dds is modified properly?
You can fix
my_coldataabove the construction ofdds.Or you can change it once it is a column of the colData:
Specifying the two levels in
contrastalso works. You should verify that you are sure of the levels and their associated counts by looking at plotCounts for individual genes. Sometimes users find that samples are mislabeled by examining plotCounts.Thanks! I checked my dds$condition before running factor and it looked like it had levels. Just to clarify, is this the correct order of events?
is step 1 even necessary if we're factoring downstream? Also if I were to relevel dds to set a different reference after step 5, would I need to run deseq again and factor after and should this be saved to a different variable like dds2 because the model changes?
Plotting the individual genes helped by the way--it confirmed what I was seeing and it ended up being that my PI had given me the data in the wrong order lol...
Again, you can factor before making dds, or you can directly factor the column as part of dds. Either works. You don’t need to do both.
Yes if you change levels you’d generally need to re-run the GLM for apeglm to work, this is discussed in the vignette under shrinkage estimators.
Thank you so much!