Entering edit mode

Hello,
I was routinely using DESeq2 for some small RNA-seq datasets, and suddenly I noticed that FDR corrections are too aggressive. After some investigation, I found out that independent filtering is wrongly determining the optimal threshold:
dds <- DESeqDataSetFromMatrix(countData = counts, colData = groups, design = ~ Group)
sizeFactors(dds) <- size_factors
dds <- DESeq(dds)
res <- results(dds)
plot(metadata(res)$filterNumRej,
type="b", ylab="number of rejections",
xlab="quantiles of filter")
lines(metadata(res)$lo.fit, col="red")
abline(v=metadata(res)$filterTheta)
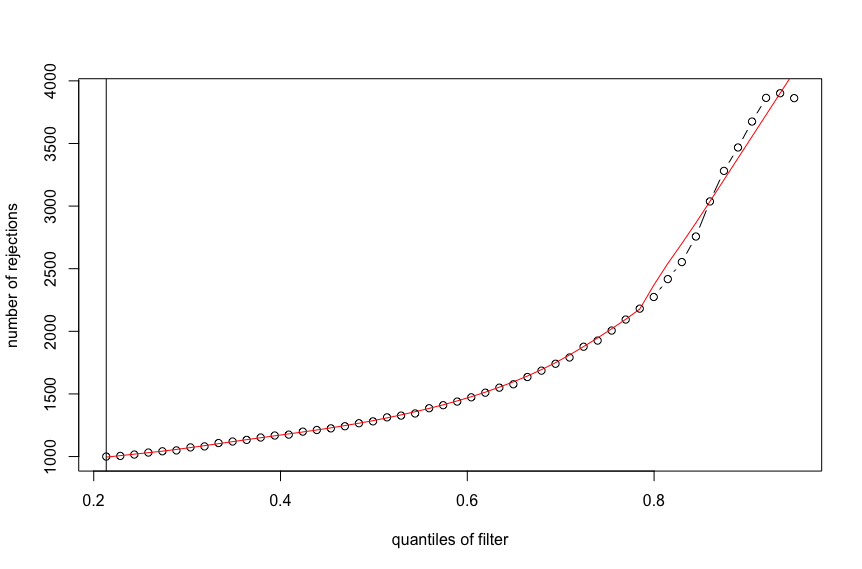
Is there any explanation of such a behavior, or I found a bug? Thanks!
sessionInfo()
R version 4.2.1 (2022-06-23)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Ventura 13.1
Matrix products: default
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] DESeq2_1.36.0 SummarizedExperiment_1.26.1 Biobase_2.56.0 MatrixGenerics_1.8.1
[5] matrixStats_0.62.0 GenomicRanges_1.48.0 GenomeInfoDb_1.32.4 IRanges_2.30.1
[9] S4Vectors_0.34.0 BiocGenerics_0.42.0
loaded via a namespace (and not attached):
[1] KEGGREST_1.36.3 genefilter_1.78.0 locfit_1.5-9.6 tidyselect_1.2.0 splines_4.2.1
[6] lattice_0.20-45 generics_0.1.3 colorspace_2.0-3 vctrs_0.5.0 utf8_1.2.2
[11] blob_1.2.3 XML_3.99-0.11 survival_3.4-0 rlang_1.0.6 pillar_1.8.1
[16] glue_1.6.2 DBI_1.1.3 BiocParallel_1.30.4 bit64_4.0.5 RColorBrewer_1.1-3
[21] GenomeInfoDbData_1.2.8 lifecycle_1.0.3 zlibbioc_1.42.0 Biostrings_2.64.1 munsell_0.5.0
[26] gtable_0.3.1 codetools_0.2-18 memoise_2.0.1 geneplotter_1.74.0 fastmap_1.1.0
[31] parallel_4.2.1 fansi_1.0.3 AnnotationDbi_1.58.0 Rcpp_1.0.9 xtable_1.8-4
[36] scales_1.2.1 cachem_1.0.6 DelayedArray_0.22.0 annotate_1.74.0 XVector_0.36.0
[41] bit_4.0.4 ggplot2_3.3.6 png_0.1-7 dplyr_1.0.10 grid_4.2.1
[46] cli_3.4.1 tools_4.2.1 bitops_1.0-7 magrittr_2.0.3 RCurl_1.98-1.9
[51] RSQLite_2.2.18 tibble_3.1.8 pkgconfig_2.0.3 crayon_1.5.2 Matrix_1.5-1
[56] httr_1.4.4 R6_2.5.1 compiler_4.2.1
Yes, there were problems with taking arg max without interpolation. It was overly sensitive to random fluctuations and resulted in extreme filtering not representing any increase in power.
I'll take a look at your example. Could you paste
metadata(res)$filterNumRejinto a comment box so I can work on it?Taking a step back, one could also say that the optimal threshold finding for independent filtering is a heuristic without clear performance properties, and should be replaced by IHW, for which mathematical guarantees exist and which should avoid this sort of problems.
Yes, I should have said, IHW is a drop in replacement, see the vignette.
Nevertheless, I should look into this default behavior and address it, I should get a chance this week.
Hello Michael,
Thank you!
Here is the output for the first figure (red line with monotonic increase):
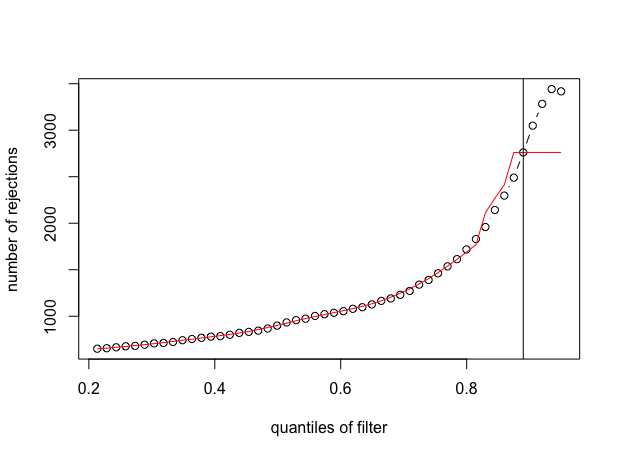
And here is the output for the second figure (red line with plateau):
Thanks for the report and for posting these examples.
I agree it's a bug and have pushed a fix to devel branch (you can obtain this easily using
devtools::install_github("mikelove/DESeq2"), v1.39.7.In terms of scope, I think this is not common. For so many quantiles of the filtering threshold, in most applications the maximum is reached in the middle, before you've tossed 98% of the features. You might benefit from doing a bit more pre-filtering of very low count features in this application anyway, and as Wolfgang said, IHW should perform better than the simple default filtering.
Nevertheless, I agree this was strange behavior for this case you presented and so I've added a check that the default filter will look to see if it can attain 90% of the fitted maximum, or 80% of the fitted maximum if not 90%. This will address these rare cases while also not changing the behavior on the vast majority of RNA-seq datasets, where you don't obtain the maximum near the end of the sequence. Typically the maximum is reached in the middle, such that looking for a point where we are close enough to the maximum in terms of the residual error works fine.
Thank you very much, Michael!