Entering edit mode

Hi all, I am trying to find which genes are different in chromatin accessibility so I used nf-core/ATAC pipeline then feed the bam files and broadpeak files output into Diffbind. I got around 100k peaks, 21k genes after annotated peaks by ChIPpeakanno, and after sorting the top genes by Fold. I got this:
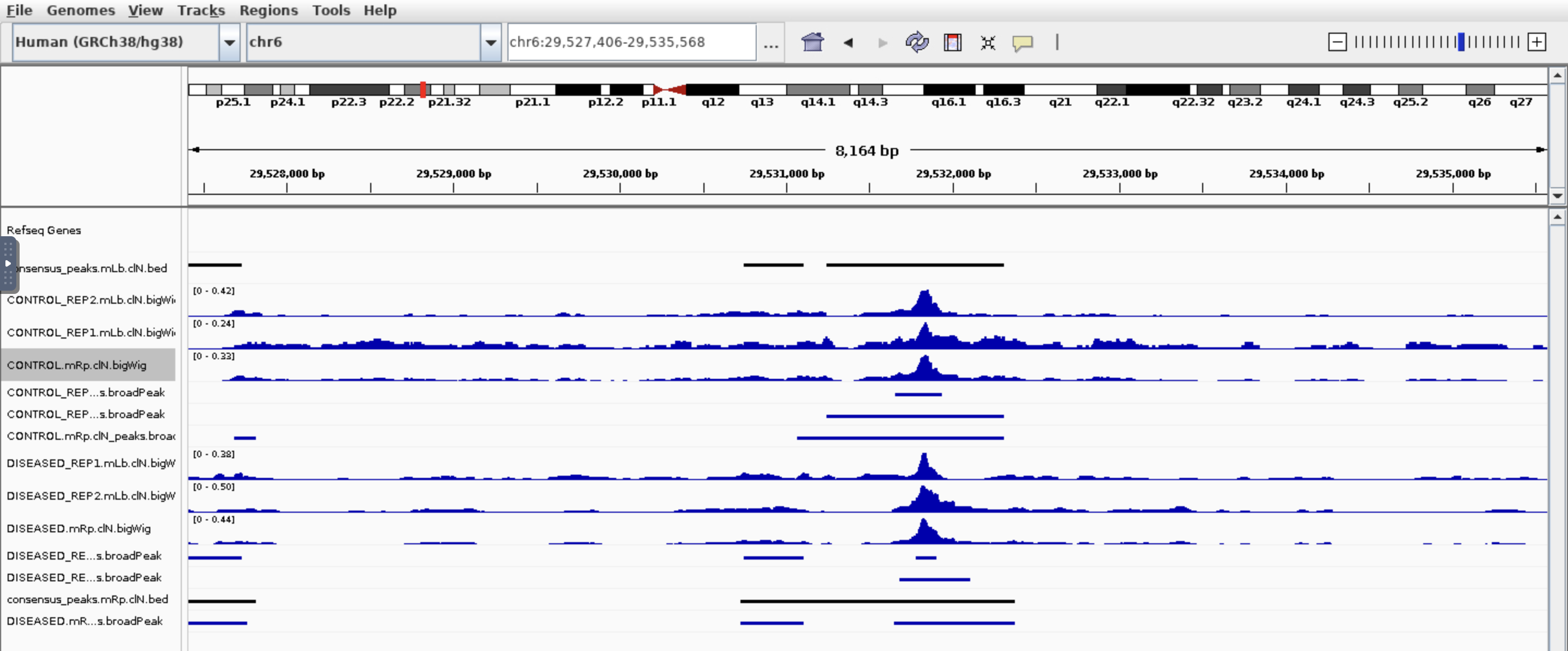
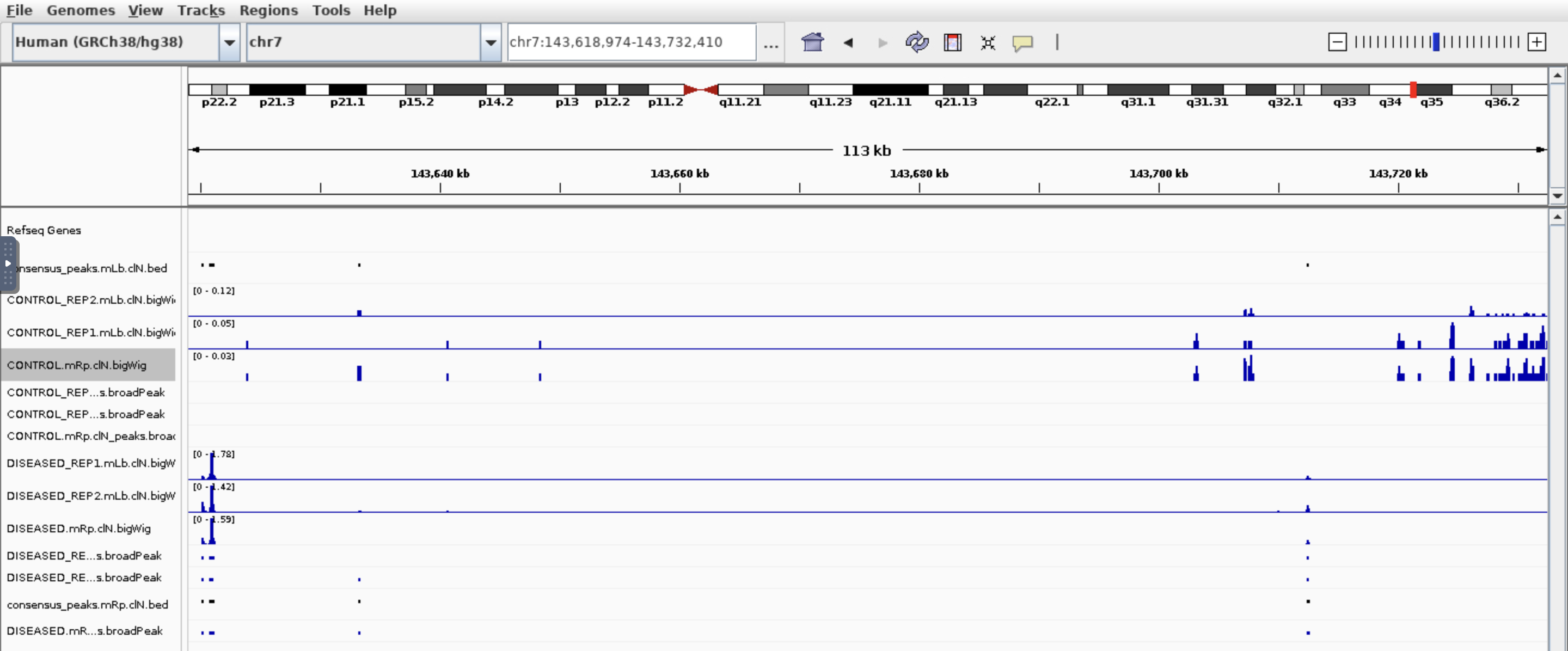
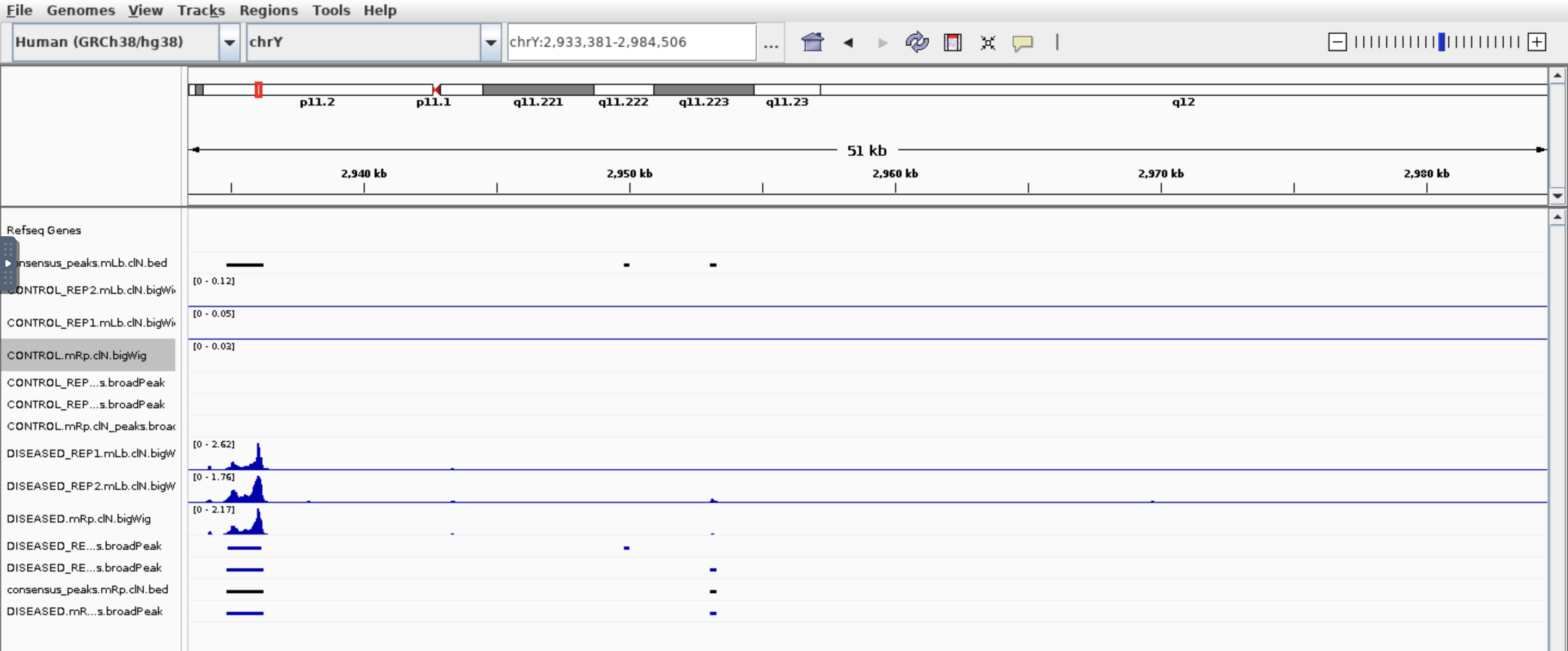
I see some genes have peaks in one condition but not other which makes sense base on the fold. But also genes have peaks in both conditions which based on the fold change is not correct. Would you please have some comments? Thank you so much!