
Hi, I'm working with whole-genome EM-seq data from an experiment with two factors:
- Two genotypes (with contrasting response to a pathogen)
- Infected and non-infected
Only one tissue and one time analyzed, with two biological replicates for each treatment combination.
I would like to know:
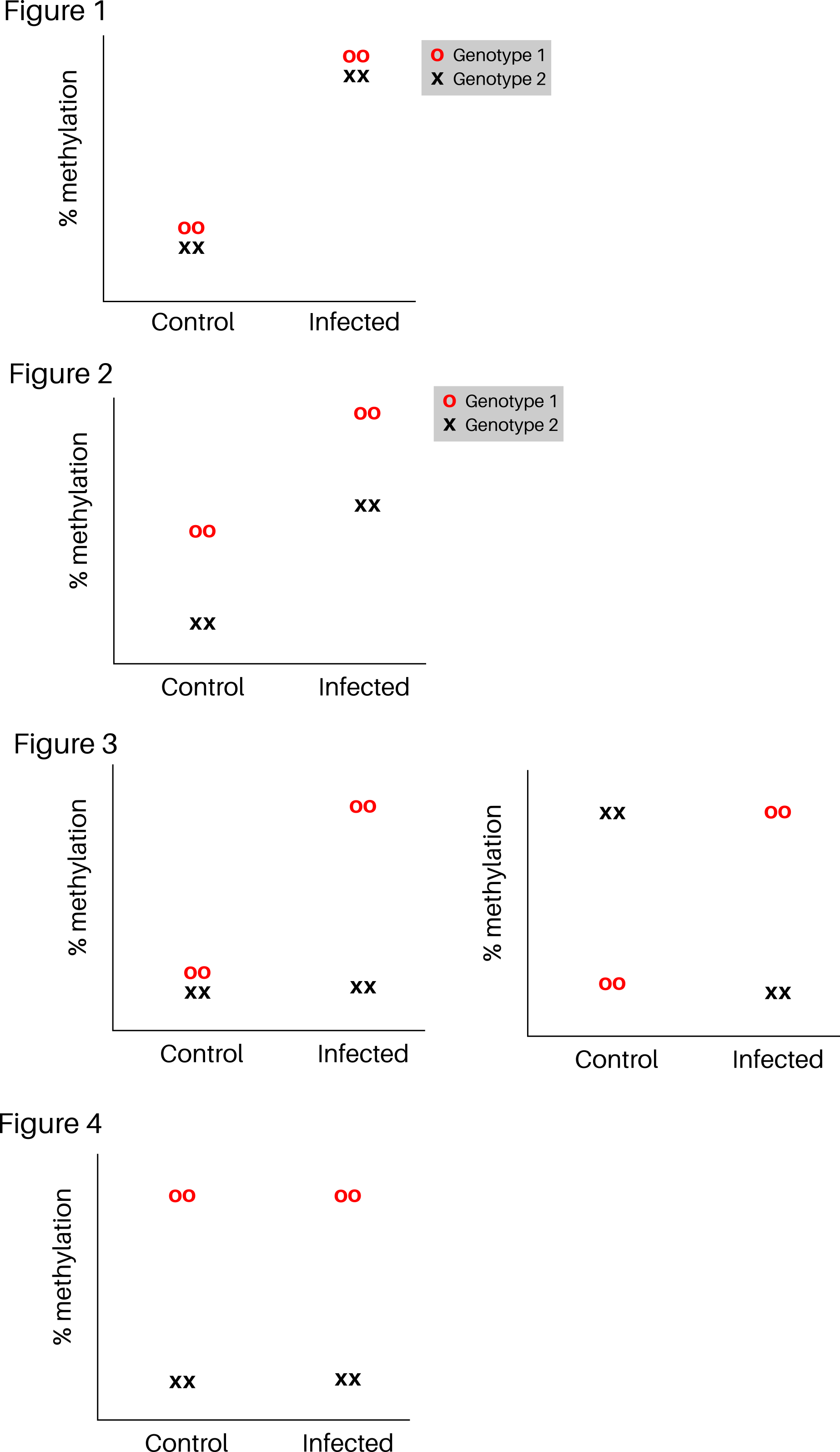
- Which sites/regions are differentially methylated in response to infection independently of genotype. (Figure 1)
- Which sites/regions are diff. methylated in response to infection and between genotypes (Figure 2)
- Which sites/regions are diff. methylated in response to infection in a genotype-dependent manner. (Figure 3)
- Which sites/regions are differentially methylated between genotypes independently of infection. (Figure 4)
I followed the Bismark alignment and extraction pipeline, and now I'm following the methylKit package tutorials.
According to it, in cases like mine with two factors and two biological replicates, I can apply logistic regressions to test for differential methylation. In the calculateDiffMeth fuction, one of the factors should be "treatment" and the other should be "covariate". I assume that to get sites diff. methylated in response to infection (goals 1-3) I need to load the infection factor as "treatment" and the genotypes as "covariate".
BUT, the resulting object is simply a list of sites/regions and their pvalue, qvalue and methdiff, without specifying the significance of each component in the regression. Which means that, for example, I can't know whether the interaction between factors was significant (case Figure 3).
My question is,
- How do I access the full information of the tested logistic regressions?
- And if I can't, how do I work around it?
I understand that to get the sites diff. methylated due to genotype (goal 4) I should switch genotype as "treatment" and infection as "covariate". But would this somehow alter the correction I should apply to the p-values?
And again, how do I identify sites diff. methylated in response to infection depending of genotype?
I thought about testing diff. meth. against infection separately for each genotype and then contrast the resulting lists, but once again, would this somehow alter the correction I must apply to the p-values?
Here's an example of my code and my results:
> meth.CHG_promoters
methylBase object with 61192 rows
--------------
chr start end strand coverage1 numCs1 numTs1 coverage2 numCs2 numTs2 coverage3 numCs3 numTs3 coverage4 numCs4 numTs4 coverage5 numCs5 numTs5 coverage6 numCs6 numTs6 coverage7 numCs7 numTs7 coverage8 numCs8 numTs8
1 HanXRQCP 97 1597 - 41775 936 40839 40096 1047 39049 49664 1041 48623 40612 628 39984 69526 3333 66193 75164 1084 74080 53468 1418 52050 53950 2110 51840
2 HanXRQCP 1266 2766 + 46631 866 45765 42949 1025 41924 54426 1054 53372 45499 669 44830 75929 3657 72272 86452 1419 85033 60449 1657 58792 59809 2511 57298
3 HanXRQCP 1524 3024 - 29216 419 28797 25039 497 24542 33037 540 32497 28064 374 27690 44816 2021 42795 53065 812 52253 37224 934 36290 35293 1300 33993
4 HanXRQCP 1876 3376 - 29126 420 28706 24639 465 24174 32690 481 32209 27573 354 27219 43789 1889 41900 53152 783 52369 37005 883 36122 34954 1186 33768
5 HanXRQCP 4681 6181 - 34258 838 33420 28101 716 27385 37988 756 37232 33004 552 32452 49461 1798 47663 64667 700 63967 45144 883 44261 42012 1227 40785
6 HanXRQCP 5495 6995 - 43559 979 42580 37327 974 36353 49203 1055 48148 42015 782 41233 64553 3094 61459 84218 1236 82982 57475 1503 55972 56022 2263 53759
--------------
sample.ids: infected_var1_r1 infected_var1_r2 control_var1_r1 control_var1_r2 infected_var2_r1 infected_var2_r2 control_var2_r1 control_var2_r2
destranded FALSE
assembly: HanXRQ-2.0
context: CHG
treament: 1 1 0 0 1 1 0 0
resolution: region
> myDiff.CHG_promoters <- calculateDiffMeth(meth.CHG_promoters,
+ covariates=data.frame(genotype=c('var1','var1','var1','var1','var2','var2','var2','var2')),
+ overdispersion = "MN",adjust="BH",mc.cores=30)
> myDiff.CHG_promoters
methylDiff object with 61192 rows
--------------
chr start end strand pvalue qvalue meth.diff
1 HanXRQCP 97 1597 - 0.9161677 1 0.196036107
2 HanXRQCP 1266 2766 + 0.9603097 1 0.089608732
3 HanXRQCP 1524 3024 - 0.9686611 1 0.108272516
4 HanXRQCP 1876 3376 - 0.9622652 1 0.163918331
5 HanXRQCP 4681 6181 - 0.8713753 1 0.134653151
6 HanXRQCP 5495 6995 - 0.9290056 1 -0.001156726
--------------
sample.ids: infected_var1_r1 infected_var1_r2 control_var1_r1 control_var1_r2 infected_var2_r1 infected_var2_r2 control_var2_r1 control_var2_r2
destranded FALSE
assembly: HanXRQ-2.0
context: CHG
treament: 1 1 0 0 1 1 0 0
resolution: region
PS1: I understand the three classic cytosine contexts (CpG, CHG and CHH) are analyzed independently from each other.
PS2: I have posted this same question in Biostars, I hope this isn't a problem https://www.biostars.org/p/9571357/#9571357
> sessionInfo( )
R version 4.1.2 (2021-11-01)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Debian GNU/Linux 11 (bullseye)
Matrix products: default
BLAS: /shared/software/lib/R/lib/libRblas.so
LAPACK: /shared/software/lib/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.utf8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] BiocParallel_1.28.3 Biobase_2.54.0 genomation_1.32.0
[4] methylKit_1.26.0 GenomicRanges_1.46.1 GenomeInfoDb_1.30.1
[7] IRanges_2.28.0 S4Vectors_0.32.4 BiocGenerics_0.40.0