
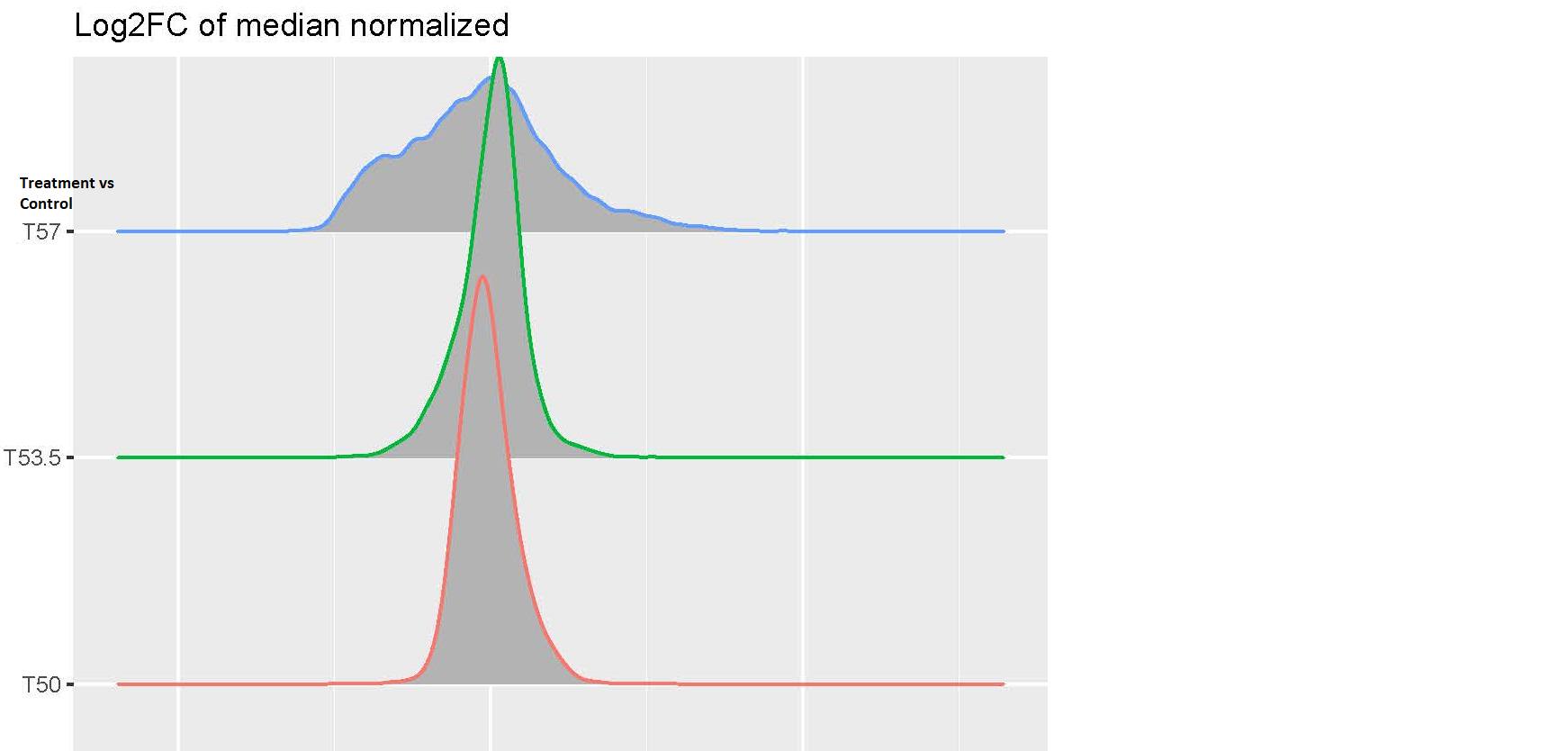
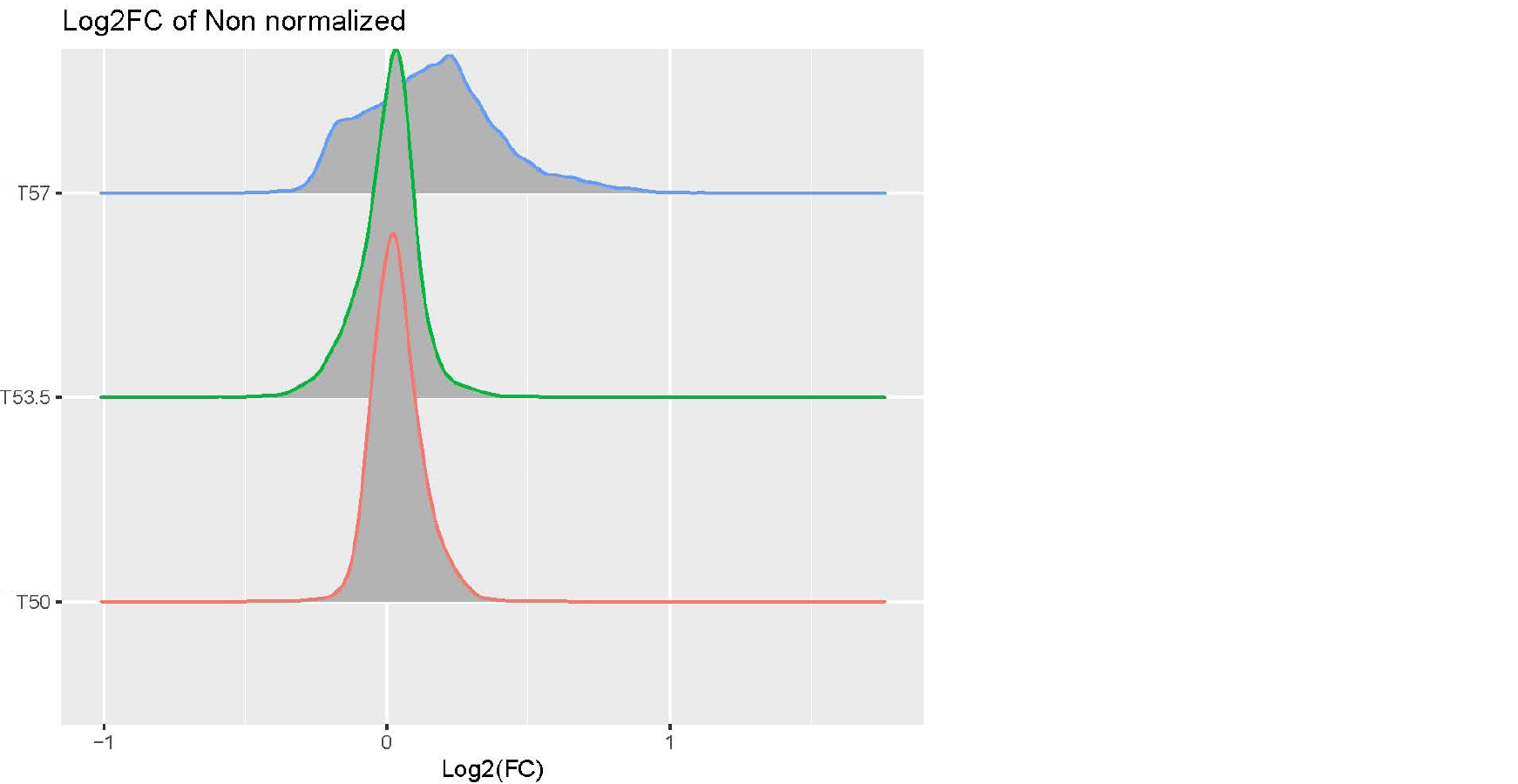
Hi, I am analyzing a data set with expression value of genes in the following conditions: cells with and without drug treatment at 3 different temperature(T): Control-T50 x3, Treatment-T53 x3, Control-T53 x3, Treatment-T53 x3, Control-T57 x3, Treatment-T57 x3. I processed the data using this pipeline: raw data > log2 transformation>median normalization for each temperature group (because temperature-dependent decrease of intensity is expected and needs to be preserved) >combine normalized samples back into one data table > limma DE analysis using one single factor matrix design (please see codes below). I then plotted log2FC of toptable output, but the logFC distribution of Treatment vs Control at T57 is very dispersed compared to other lower temperatures(see images attached). I've attached the logFC plots and intensity plots for non-normalized (raw) and median-normalized data set (please note the temperature-dependent decrease of intensity is expected).
My questions are (1) should I worry about this issue? (2) how can I fix this? I suspected the high variability of T57 samples but it doesn't seem so from the intensity plots. Any input would be appreciated, thanks very much. Please let me know if any other information I need to provide.


groups <- factor(groups, levels = c("ControlT50",
"TreatmentT50",
"ControlT53",
"TreatmentT53",
"ControlT57",
"TreatmentT57"
))
design <- model.matrix(~ 0 + groups)
colnames(design) <- gsub("groups", "", colnames(design))
rownames(design) <- colnames(limma_df)
fitall <- limma::lmFit(limma_df, design)
meanint <- apply(limma_df, 1, function (x) {mean(x, na.rm =T)})
fitall$Amean <- meanint
contrast.matrix <- makeContrasts(TreatmentT50-ControlT50,
TreatmentT53.5-ControlT53,
TreatmentT57-ControlT57,
levels=design)
fitall <- limma::contrasts.fit(fitall, contrast.matrix)
fitall_eb <- limma::eBayes(fitall, trend = T)
toptable <- data.frame()
for (i in 1:length(colnames(fitall_eb))) {
toptabdf <- limma::topTable(fitall_eb, coef = colnames(fitall_eb)[i],number = Inf, confint = T, adjust.method="BH")
toptabdf <- toptabdf %>% mutate (Comparison = colnames(fitall_eb)[i])
toptable <- rbind(toptable, toptabdf)
}
toptable %>%
ggplot( aes(x = logFC, y = Comparison, color = Comparison ) ) +
ggridges::geom_density_ridges()
*limma* version: 3.65.2
# include your problematic code here with any corresponding output
# please also include the results of running the following in an R session
sessionInfo( R version 4.3.1 (2023-06-16 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 17763))


Is this perhaps a thermal protein profiling experiment?
Yes, it is! I didn't explain it clearly in my 1st post. This isn't the classic way to design a thermal profiling experiment which typically uses a wide temperature range, but due to the limited multiplexing power of mass spectrometry proteomics, we are trading temperature points for more biological replicates. We are trying to find the optimal matrix design to answer the following questions: (1) what proteins show differential abundance in Treatment vs Control at one temperature? (2) how can we use the temperature to improve the differential expression results? For now, we are using the single-factor design and make contrasts of Treatment vs Control at each temperature (as said in my post, we also consider looking at the effect of temperature: e.g. (Treatment - Control at T57) - (Treatment - Control at T50)). But now we get ~2,000 hits at T57 of Treatment vs Control (adj.P < 0.1), but no hits at T53 and T50, that made me concerned of false positives at T57. I wonder if there are flaws in my analysis pipeline that inflates the hits. I've attached the Pvalue histogram of T57 TreatvsCon if that's helpful. Thank you very much for looking into this.