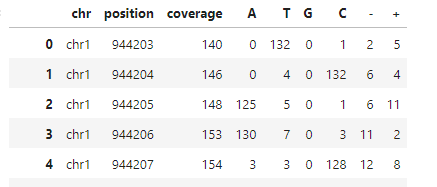
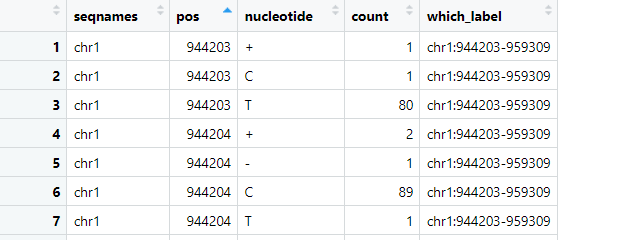
Entering edit mode

I have mRNA sequencing data that I've aligned to a genome. Specifically, I am interested in determining the total number of reads at each base and the percentage of occurrences of A, T, G, C, deletions, and insertions at these bases. I have utilized both pysam pileup and Rsamtools pileup to achieve this. However, I noticed that their outputs are inconsistent, even though I used the same BAM file as input for both tools. Below is the code I implemented:
Pysam:
bamfile = AlignmentFile(bam_dir, "rb")
pileups = bamfile.pileup('chr1', 944202, 959308, truncate=True, min_mapping_quality=20)
columns=['chr', 'position', "coverage", 'A', 'T', 'G', 'C', '-', '+']
df = pd.DataFrame(columns=columns)
for pileupcolumn in pileups:
data = []
for pileupread in pileupcolumn.pileups:
if pileupread.is_del:
data.append('-')
elif pileupread.indel:
data.append('+')
else:
data.append(pileupread.alignment.query_sequence[pileupread.query_position])
base_counts = Counter(data)
row = {
'chr': 'chr1',
'position': pileupcolumn.pos+1,
'coverage': pileupcolumn.n,
'A': base_counts['A'],
'T': base_counts['T'],
'G': base_counts['G'],
'C': base_counts['C'],
'-': base_counts['-'],
'+': base_counts['+']
}
df = df.append(row, ignore_index=True)
Rsamtools
param <- ScanBamParam(which=GRanges(strand = "-",
seqnames = "chr1",
ranges = IRanges(start=944203,
end=959309)))
pilup_params = Rsamtools::PileupParam(max_depth = 102500,
min_mapq = 20,
distinguish_nucleotides = T,
ignore_query_Ns = F,
include_insertions = T,
distinguish_strands = F)
bam.pileup = pileup(bamfile,
scanBamParam=param,
pileupParam = pilup_params)
bam.pileup = bam.pileup[order(bam.pileup$pos), ]
The read count outputs from pysam and Rsamtools vary significantly, as can be seen in the attached pictures. Have I made any critical errors in my approach?