Entering edit mode

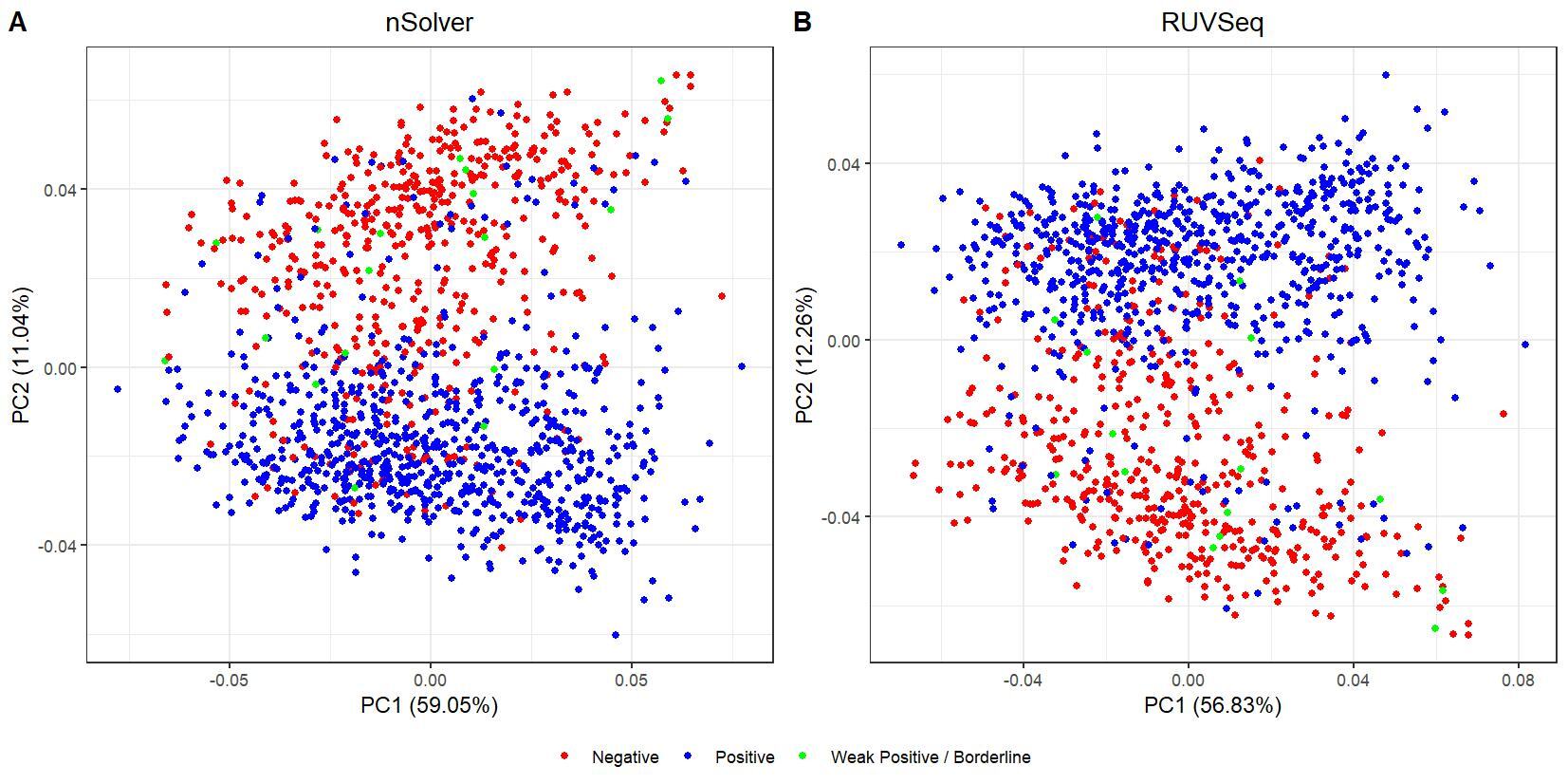
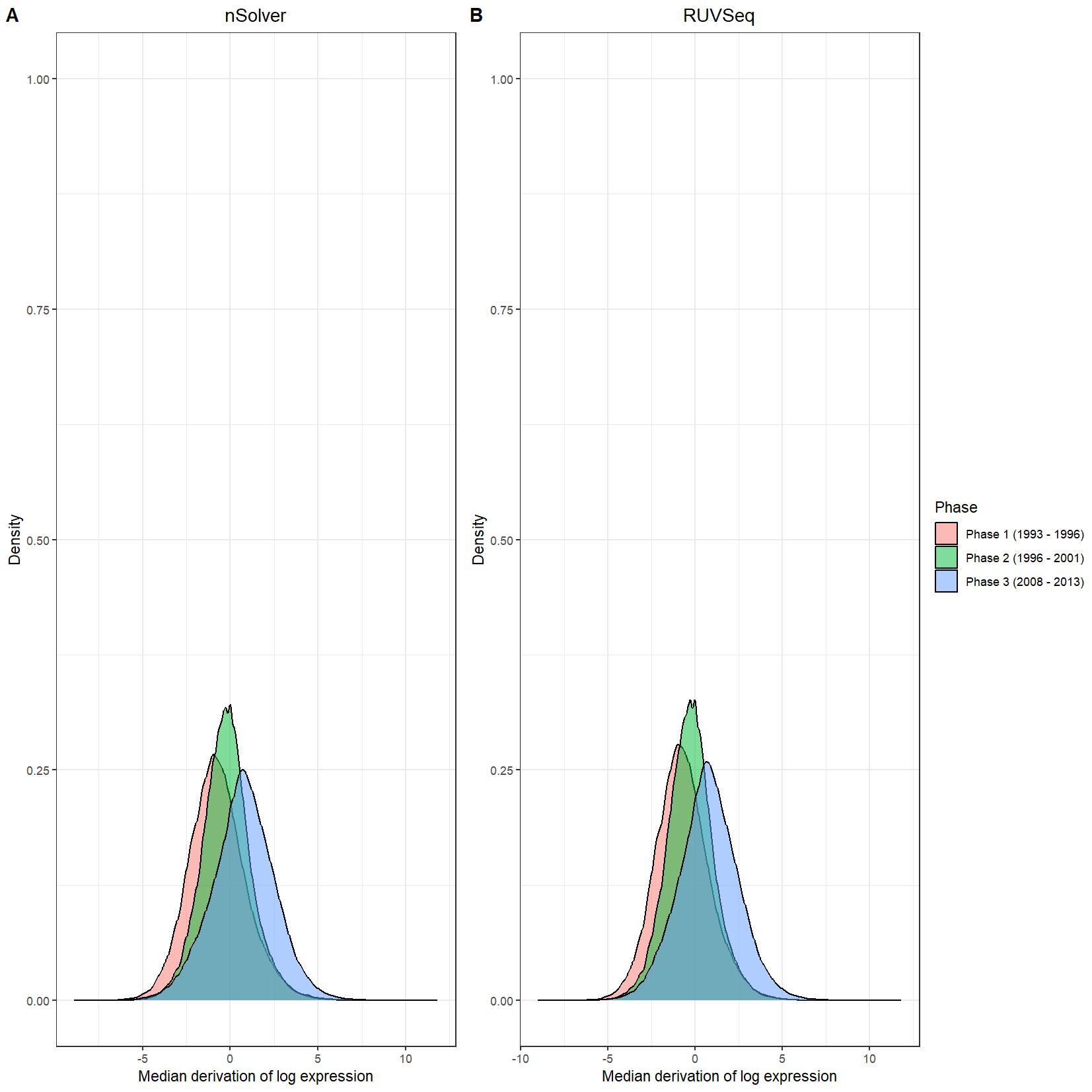
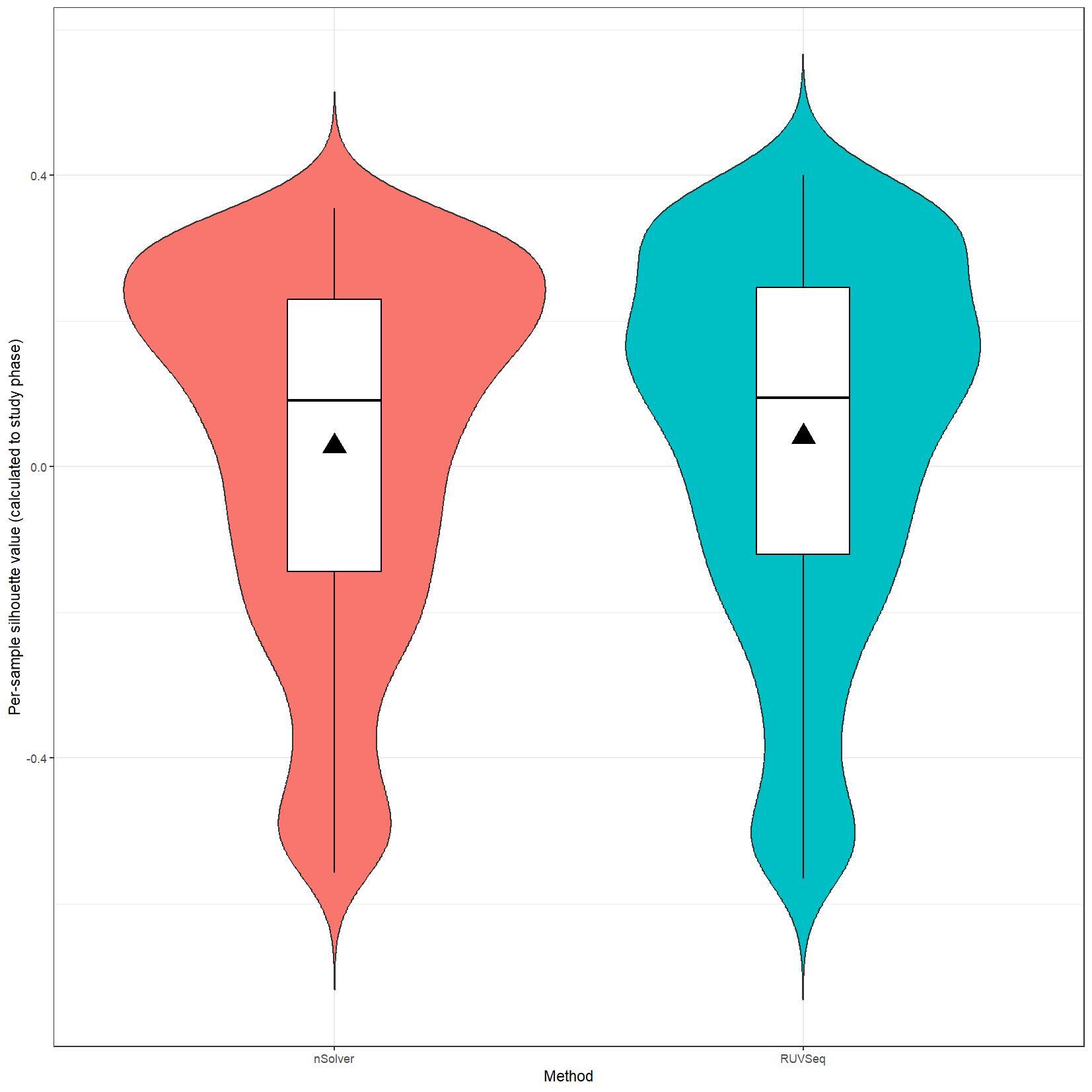
We have been trying to replicate part of the analysis of the paper titled 'An approach for normalization and quality control for NanoString RNA expression data' by Bhattacharya et al. To get a feeling for the effect of RUVseq normalization compared to nSolver normalization we tried to replicate some of the plots from Figure 2 using the CBCS data from GEO GSE148426 (which includes GSE148425 and GSE148418). At the moment, we are failing achieve similar PCA plots using the already normalized data in GEO. Most of the biological variance goes in PC2. Also, our silhouette plots and density plots differ a little. Could you be so kind to help us understand what we are doing wrong? You can find the code and images below. Thank you for your help!
######## LOAD OR INSTALL NECESSARY PACKAGES ########
library(nanostringr)
library(ggpubr)
library(tidyverse)
library(cluster)
library(ggplot2)
library(ggfortify)
######## DEFINE GLOBAL VARIABLES ########
NSOLVER.DATA.FOLDER <- "./GSE148418/" # Directory containing the RCC files downloaded from GEO
RUVSEQ.DATA.FOLDER <- "./GSE148425/" # Directory containing the RCC files downloaded from GEO
RESULTS.FOLDER <- "./Results/"
NSOLVER.ANNOTATION.FILE <- "./GSE148418/GSE148418_metadata.csv" # Annotation file generated from raw series matrix
RUVSEQ.ANNOTATION.FILE <- "./GSE148425/GSE148425_metadata.csv" # Annotation file generated from raw series matrix
PLOT.COLORS <- c("red", "blue", "green", "orange", "darkgreen", "black", "yellow", "pink")
# Read data
rcc_files_nsolver <- read_rcc(NSOLVER.DATA.FOLDER)
nsolver_dat <- rcc_files_nsolver$raw
rownames(nsolver_dat) <- nsolver_dat$Name
rcc_files_ruvseq <- read_rcc(RUVSEQ.DATA.FOLDER)
ruvseq_dat <- rcc_files_ruvseq$raw
rownames(ruvseq_dat) <- ruvseq_dat$Name
clin_lab_nsolver <- read.csv(NSOLVER.ANNOTATION.FILE) # Import Annotations
rownames(clin_lab_nsolver) <- paste(clin_lab_nsolver$Sample_geo_accession, clin_lab_nsolver$Sample_description, sep="_")
clin_lab_nsolver$Sample_characteristics_ch1 <- gsub("study phase: ", "", clin_lab_nsolver$Sample_characteristics_ch1)
clin_lab_nsolver$Sample_characteristics_ch1.2 <- gsub("estrogen receptor status: ", "", clin_lab_nsolver$Sample_characteristics_ch1.2)
clin_lab_ruvseq <- read.csv(RUVSEQ.ANNOTATION.FILE) # Import Annotations
rownames(clin_lab_ruvseq) <- paste(clin_lab_ruvseq$Sample_geo_accession, clin_lab_ruvseq$Sample_description, sep="_")
clin_lab_ruvseq$Sample_characteristics_ch1.1 <- gsub("study phase: ", "", clin_lab_ruvseq$Sample_characteristics_ch1.1)
clin_lab_ruvseq$Sample_characteristics_ch1.2 <- gsub("estrogen receptor status: ", "", clin_lab_ruvseq$Sample_characteristics_ch1.2)
# Log transform data
log_nsolver_dat <- log2(nsolver_dat[,-c(1:3)]+1)
log_ruvseq_dat <- log2(ruvseq_dat[,-c(1:3)]+1)
# Density plots log data
nsolver_medians <- apply(as.data.frame(log_nsolver_dat), 1, median)
nsolver_devs_from_median <- as.data.frame(log_nsolver_dat) - nsolver_medians
nsolver_devs_from_median_long <- gather(nsolver_devs_from_median, key = patient, value = value)
nsolver_devs_from_median_long$Phase <- clin_lab_nsolver[nsolver_devs_from_median_long$patient, 'Sample_characteristics_ch1']
ruvseq_medians <- apply(as.data.frame(log_ruvseq_dat), 1, median)
ruvseq_devs_from_median <- as.data.frame(log_ruvseq_dat) - ruvseq_medians
ruvseq_devs_from_median_long <- gather(ruvseq_devs_from_median, key = patient, value = value)
ruvseq_devs_from_median_long$Phase <- clin_lab_ruvseq[ruvseq_devs_from_median_long$patient, 'Sample_characteristics_ch1.1']
nsolver_density_plot <- ggplot(nsolver_devs_from_median_long, aes(x = value, fill = Phase)) +
geom_density(alpha = 0.5) +
scale_color_manual(values=PLOT.COLORS) +
theme_bw() +
labs(x = "Median derivation of log expression", y = "Density") +
ggtitle("nSolver") +
theme(plot.title = element_text(hjust = 0.5)) +
scale_y_continuous(limits = c(0, 1))
ruvseq_density_plot <- ggplot(ruvseq_devs_from_median_long, aes(x = value, fill = Phase)) +
geom_density(alpha = 0.5) +
scale_color_manual(values=PLOT.COLORS) +
theme_bw() +
labs(x = "Median derivation of log expression", y = "Density") +
ggtitle("RUVSeq") +
theme(plot.title = element_text(hjust = 0.5))+
scale_y_continuous(limits = c(0, 1))
jpeg(paste0(RESULTS.FOLDER, "Density_plots.jpeg"), units="cm", width=28, height=28, res=150)
ggarrange(nsolver_density_plot, ruvseq_density_plot, common.legend = TRUE, legend="right", labels = LETTERS[1:2])
dev.off()
# Silhouette violin plots
clin_lab_nsolver$Silhouette.cluster <- clin_lab_nsolver$Sample_characteristics_ch1
clin_lab_nsolver <- clin_lab_nsolver %>% mutate(Silhouette.cluster = recode(Silhouette.cluster, "Phase 1 (1993 - 1996)" = 1, "Phase 2 (1996 - 2001)" = 2, "Phase 3 (2008 - 2013)" = 3))
nsolver_silhouette_vals <- silhouette(clin_lab_nsolver[colnames(nsolver_dat[,-c(1:3)]), 'Silhouette.cluster'], dist(t(log_nsolver_dat))^2)
clin_lab_ruvseq$Silhouette.cluster <- clin_lab_ruvseq$Sample_characteristics_ch1.1
clin_lab_ruvseq <- clin_lab_ruvseq %>% mutate(Silhouette.cluster = recode(Silhouette.cluster, "Phase 1 (1993 - 1996)" = 1, "Phase 2 (1996 - 2001)" = 2, "Phase 3 (2008-2013)" = 3))
ruvseq_silhouette_vals <- silhouette(clin_lab_ruvseq[colnames(ruvseq_dat[,-c(1:3)]), 'Silhouette.cluster'], dist(t(log_ruvseq_dat))^2)
silhouette_vals_df <- rbind(as.data.frame.matrix(nsolver_silhouette_vals), as.data.frame.matrix(ruvseq_silhouette_vals))
silhouette_vals_df$Method <- c(rep("nSolver", nrow(nsolver_silhouette_vals)), rep("RUVSeq", nrow(ruvseq_silhouette_vals)))
jpeg(paste0(RESULTS.FOLDER, "Silhouette_plots.jpeg"), units="cm", width=28, height=28, res=150)
ggplot(silhouette_vals_df, aes(x = Method, y = sil_width, fill = Method)) +
geom_violin(trim=F) +
geom_boxplot(width=0.2, color="black", fill="white") +
stat_summary(fun.y=mean, geom="point", shape=17, size=6, fill="black") +
scale_color_manual(values=PLOT.COLORS) +
theme_bw() +
theme(legend.position = "none") +
labs(x = "Method", y = "Per-sample silhouette value (calculated to study phase)")
dev.off()
# PCA plots
pca_nsolver_dat <- log_nsolver_dat[,clin_lab_nsolver$Sample_characteristics_ch1.2 != "Unknown"] # remove samples with unknown subtype to make figure more clear
pca_ruvseq_dat <-log_ruvseq_dat[,clin_lab_ruvseq$Sample_characteristics_ch1.2 != "Unknown"]# remove samples with unknown subtype to make figure more clear
pca_nsolver <- prcomp(t(pca_nsolver_dat), scale. = F)
pca_plot_nsolver <- autoplot(pca_nsolver,
data = clin_lab_nsolver[clin_lab_nsolver$Sample_characteristics_ch1.2 != "Unknown",],
colour = "Sample_characteristics_ch1.2",
label = F) + scale_color_manual(values=PLOT.COLORS) + theme_bw() + ggtitle("nSolver") + theme(plot.title = element_text(hjust = 0.5), legend.title = element_blank())
pca_ruvseq <- prcomp(t(pca_ruvseq_dat), scale. = F)
pca_plot_ruvseq <- autoplot(pca_ruvseq,
data = clin_lab_ruvseq[clin_lab_ruvseq$Sample_characteristics_ch1.2 != "Unknown",],
colour = "Sample_characteristics_ch1.2",
label = F) + scale_color_manual(values=PLOT.COLORS) + theme_bw() + ggtitle("RUVSeq") + theme(plot.title = element_text(hjust = 0.5), legend.title = element_blank())
jpeg(paste0(RESULTS.FOLDER, "PCA_plots_estrogen_receptor_status.jpeg"), units="cm", width=28, height=14, res=150)
ggarrange(pca_plot_nsolver, pca_plot_ruvseq, common.legend = TRUE, legend="bottom", labels = c("A", "B"))
dev.off()

From the RCC files I downloaded from GEO I get the following values after loading them in RStudio and pulling the expression values for patient 1:
So that is not the same data as expected... They were obtained from the GSE148426_RAW.tar file from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148426.
It is odd though because if I pull the data in RStudio directly using GEOquery, I do get the same values as were expected by you for the GSM4468667 and GSM4469975 entries. I will try to generate the figures again and will let you know if I retrieve different results.
I believe we mixed up the raw data with the nSolver and RUVseq normalized data. After importing the data using GEOquery the PCA and density plots seem identical to the figures from your paper. The only figures that seem off are the silhouette plots. We are wondering how they were generated as we are not familiar with creating these type of plots but they seem to capture valuable information. Do you happen to know how they were generated for your paper?
We are still unable to reproduce the silhouette plots. Do you happen to know how these were generated? We applied the silhouette function from the R cluster package using the study phase as cluster label (see below). Should we use something different?
It also seems as if the nSolver and RUVSeq data are already log-transformed, especially since the RUVSeq data contains negative values. Do you know if this holds true and which log (e.g. 10, 2, n) was used? We could not find it in the GEO descriptions.
Thank you in advance!
VST data was used (see the code repo for the paper). This is like a logarithm.
I believe we used standard silhouette function.