Entering edit mode

Dear all,
I have the following issue: I have a Seurat object with 7 layers of raw gene expression counts for 7 different patients.
In order to annotate the object with SingleR I have now used the function "JoinLayers" to combine all counts into one layer of counts of all patients together. I was able to annotate the clusters of cells with
kid.filtered_new<- JoinLayers(kid.filtered)
pbmc_counts_combined <- GetAssayData(kid.filtered_new, slot = 'counts')
pred_combined <- SingleR(test = pbmc_counts_combined,
ref = ref,
labels = ref$label.main)
kid.filtered_new$singleR.labels <- pred_combined$labels[match(rownames(kid.filtered_new@meta.data), rownames(pred_combined))]
DimPlot(kid.filtered, reduction = 'umap', group.by = 'singleR.label`s')
However, I would like to use for the annotation not the labels of every cell. Instead I would like to use for the annotation the seurat clusters:
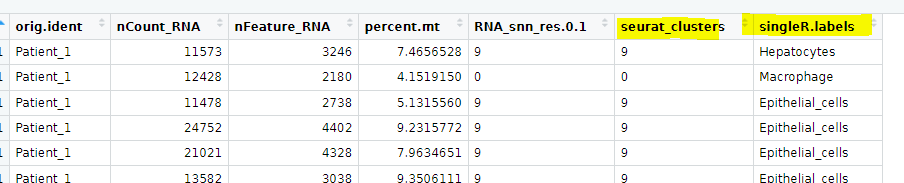
I am able to clusters them by Seurat clusters but the annotation via SingleR was done per single cell. How can I get the annotation done per seurat clusters?
DimPlot(kid.filtered, reduction = 'umap', group.by = 'singleR.labels')
I tried this but it is giving me an error:
pred_combined <- SingleR(test = pbmc_counts_combined,
ref = ref,
labels = ref$label.main, clusters='seurat_clusters')
Error in compute_ugroup(group, ncol(x), reorder) :
incorrect length for 'group'
Thank you for any input!
Bine
Please ask at biostars.org for Seurat help which is not a Bioconductor package. As said, SingleR is generic as it can start from a matrix and label vector, that here is a Seurat problem.
Ok thanks, done. Shall I delete this post?
Your post is very helpful.