Entering edit mode

How do I ensure that filtration of lowly expressed counts in RNA seq data was done correctly? my goal is to find DEGs using limma voom This is a plot of data before and after filtering by filterbyExpr ?
Code should be placed in three backticks as shown below
cpm <- cpm(dge)
lcpm <- cpm(dge, log=TRUE)
L <- mean(dge$samples$lib.size) * 1e-6
M <- median(dge$samples$lib.size) * 1e-6
c(L, M)
lcpm.cutoff <- log2(10/M + 2/L)
library(RColorBrewer)
nsamples <- ncol(dge$counts)
col <- brewer.spectral(nsamples)
par(mfrow=c(1,2))
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.55), las=2, main="", xlab="")
title(main="A. Raw data", xlab="Log-cpm")
abline(v=lcpm.cutoff, lty=3)
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
##FilterbyExpr
keep.exprs <- filterByExpr(dge, group = dge$samples$group)
dge <- dge[keep.exprs,, keep.lib.sizes=FALSE]
dim(dge)
lcpm <- cpm(dge, log=TRUE)
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.5), las=2, main="", xlab="")
title(main="B. Filtered data", xlab="Log-cpm")
abline(v=lcpm.cutoff, lty=3)
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}

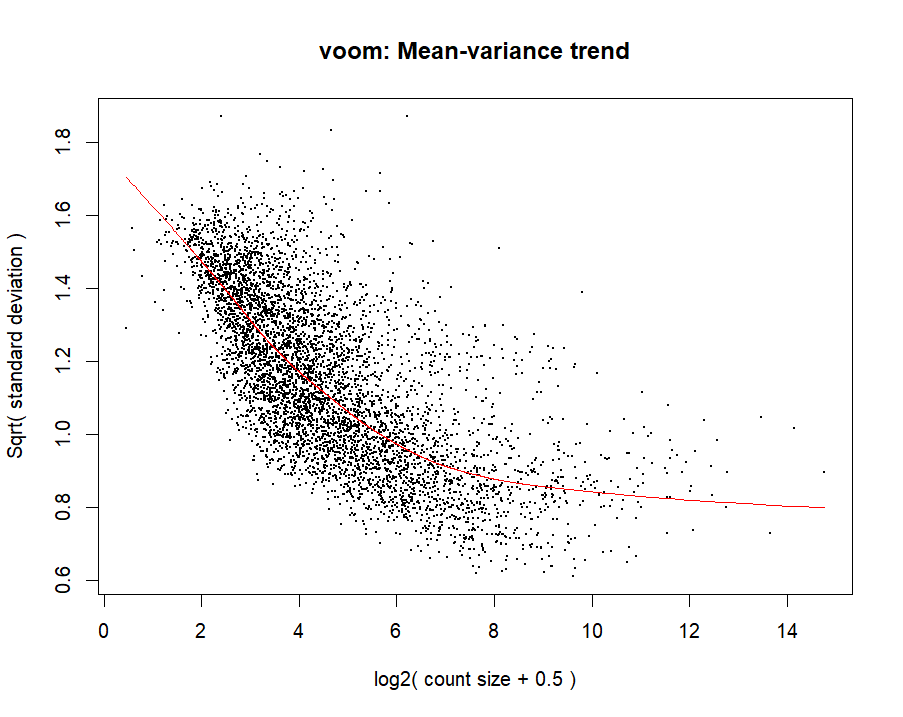
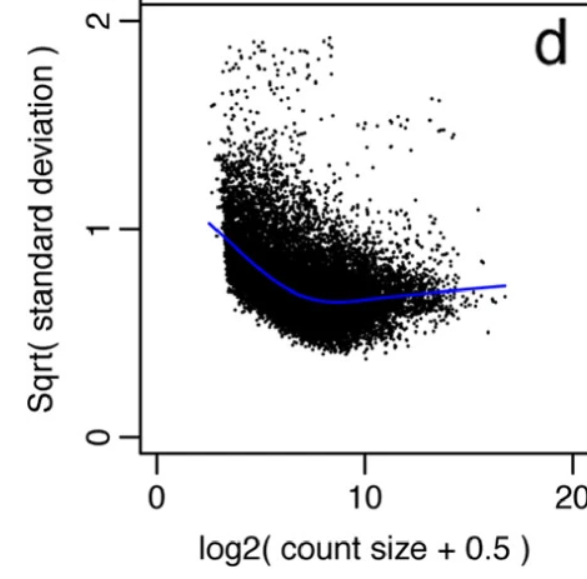
Cross-posted to Biostars https://www.biostars.org/p/9584889/