
Hello everybody,
I am analyzing a dataset of RNAseq data generated previously. Basically, the dataset is made of 3 conditions, with 4 biological replicates in each condition, and 2 technical replicates per biological condition (maybe the best choice, but the data is here...). I am observing massive LFC shrinkage (almost all genes LFC are shrinked to 0) in my analysis, even though the counts seems OK.
I first filter the counts using counts_data <- counts_data[which(rowSums(counts_data>=10) >= 8),], ending up with 15126 genes (that might be important?).
I create the dds object using dds <- DESeqDataSetFromMatrix(countData = counts_data, colData = metadata, design = ~Experimental_condition) and then do ddsColl <- collapseReplicates(dds, dds$Replicate, dds$Sample_name) to merge technical replicates, and proceed with the analysis normally:
contrast_exchange_vs_alone <- c("Experimental_condition", "E", "B")
res_exchange_vs_alone <- results(ddsColl, contrast=contrast_exchange_vs_alone, alpha = 0.05)
res_exchange_vs_alone_lfcshrink <- lfcShrink(ddsColl, coef="Experimental_condition_E_vs_B", res=res_exchange_vs_alone) #type='apeglm' default
Here is the MA plot before shrinkage:
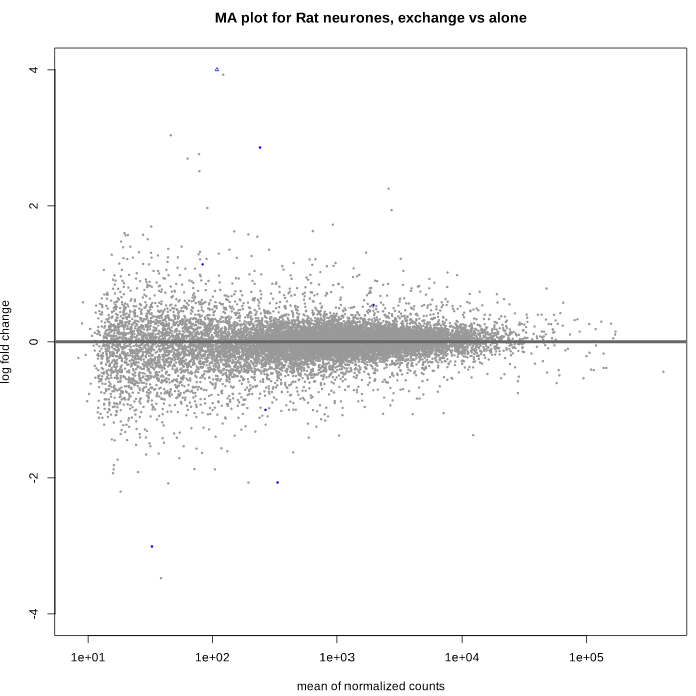
And after shrinkage (apeglm):
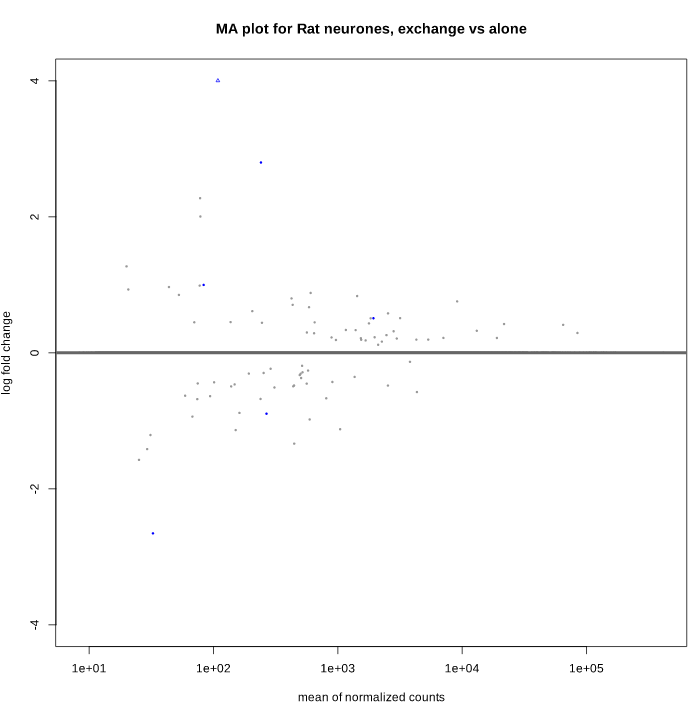
Here is the volcano plot after shrinkage:
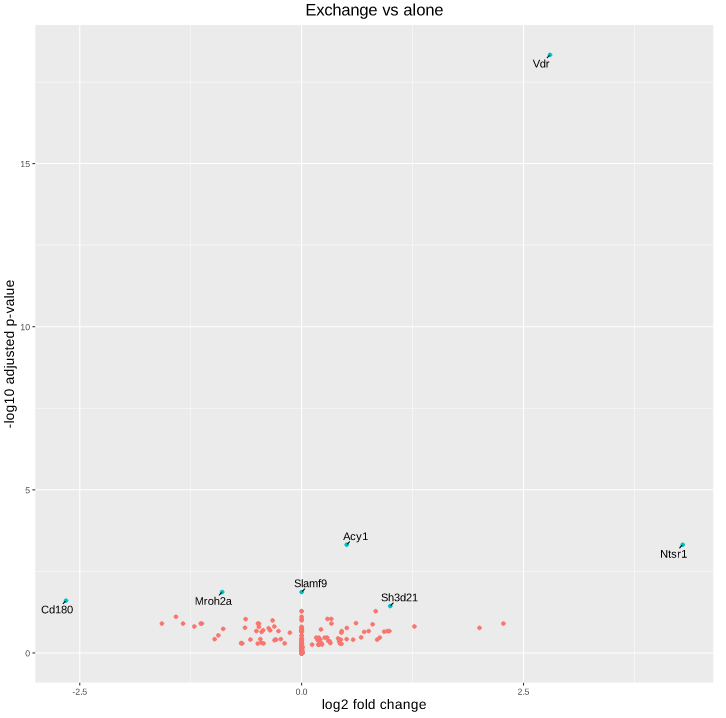
As you can see, some gene's LFC are heavily shifted towards 0. Let's take a closer look at gene ENSRNOG00000008045 (a.k.a Slamf9), here is the plotCounts for normalized counts:
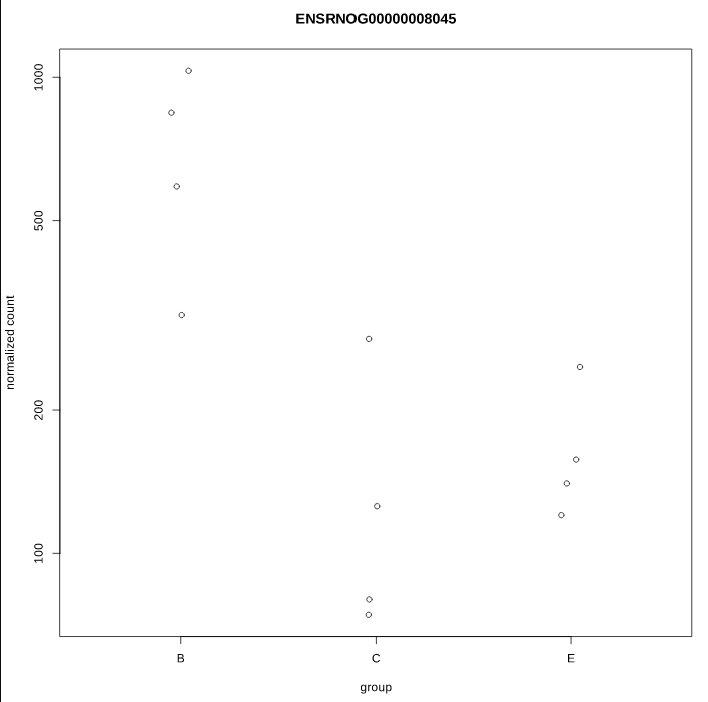
Same gene, raw counts:
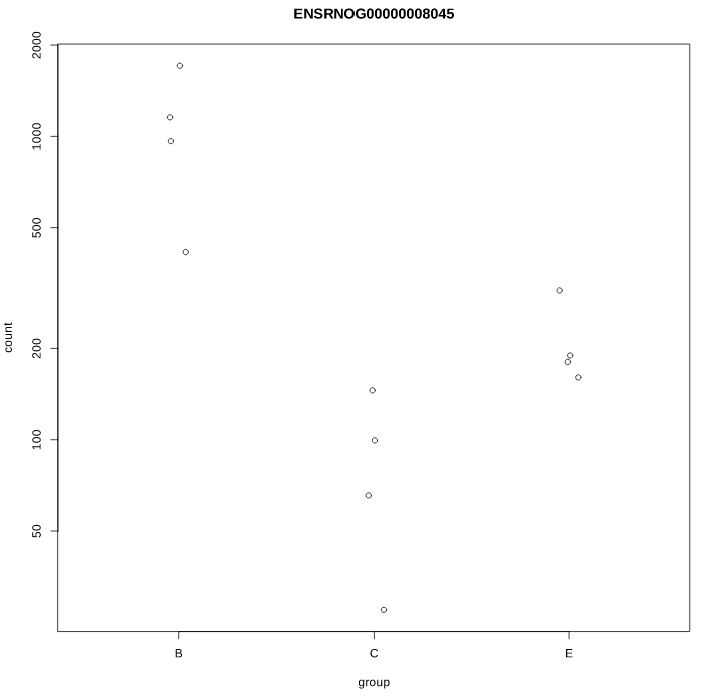
There is indeed some variability between replicates, but I am surprised that the LFC was shrinked to 0 for this gene. All counts are > 100 for the conditions under study ("E" and "B"). Do you have any clue why LFC shrink is so drastic, and what is going on in my analysis/dataset?
Thank you in advance for your help and insights,
Theo
Edit 1:
The MA plot looks similar with lfcShrink(type='ashr'):
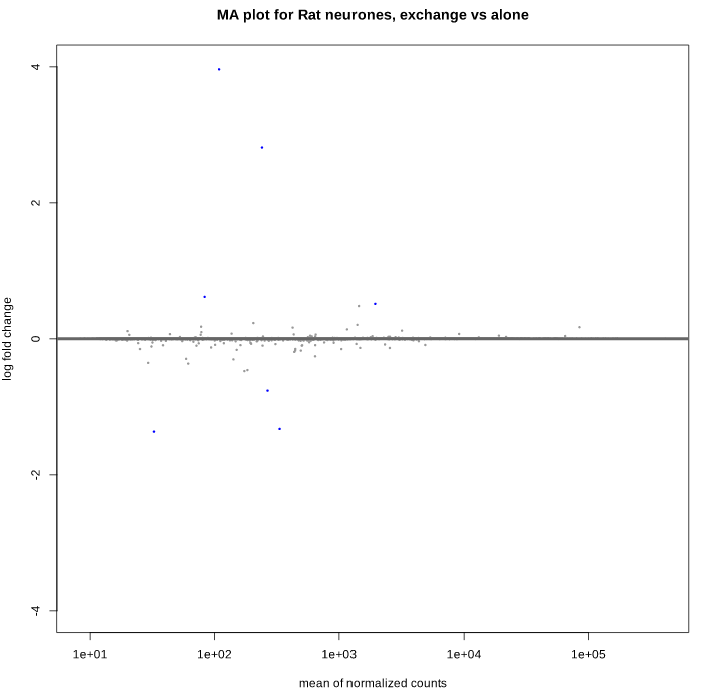
I suspect something is off with the data but I have not been able to track it down...
> sessionInfo()
R version 4.3.2 (2023-10-31)
Platform: x86_64-conda-linux-gnu (64-bit)
Running under: Ubuntu 20.04.5 LTS
Matrix products: default
BLAS/LAPACK: /home/xxxx/miniconda3/envs/yyyy/lib/libopenblasp-r0.3.25.so; LAPACK version 3.11.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
time zone: Europe/Paris
tzcode source: system (glibc)
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] org.Rn.eg.db_3.18.0 org.Hs.eg.db_3.18.0
[3] Orthology.eg.db_3.18.0 AnnotationDbi_1.64.1
[5] ggrepel_0.9.4 pheatmap_1.0.12
[7] GGally_2.2.0 DEGreport_1.38.4
[9] ggiraph_0.8.8 ggplot2_3.4.4
[11] DESeq2_1.42.0 SummarizedExperiment_1.32.0
[13] Biobase_2.62.0 MatrixGenerics_1.14.0
[15] matrixStats_1.2.0 GenomicRanges_1.54.1
[17] GenomeInfoDb_1.38.1 IRanges_2.36.0
[19] S4Vectors_0.40.2 BiocGenerics_0.48.1
[21] FactoMineR_2.9 BiocManager_1.30.22
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3 ggdendro_0.1.23
[3] shape_1.4.6 magrittr_2.0.3
[5] estimability_1.4.1 farver_2.1.1
[7] GlobalOptions_0.1.2 zlibbioc_1.48.0
[9] vctrs_0.6.5 memoise_2.0.1
[11] RCurl_1.98-1.13 htmltools_0.5.7
[13] S4Arrays_1.2.0 progress_1.2.3
[15] broom_1.0.5 SparseArray_1.2.2
[17] htmlwidgets_1.6.4 plyr_1.8.9
[19] emmeans_1.9.0 cachem_1.0.8
[21] uuid_1.1-1 lifecycle_1.0.4
[23] iterators_1.0.14 pkgconfig_2.0.3
[25] Matrix_1.6-4 R6_2.5.1
[27] fastmap_1.1.1 GenomeInfoDbData_1.2.11
[29] clue_0.3-65 numDeriv_2016.8-1.1
[31] digest_0.6.33 colorspace_2.1-0
[33] reshape_0.8.9 RSQLite_2.3.4
[35] labeling_0.4.3 fansi_1.0.6
[37] mgcv_1.9-1 httr_1.4.7
[39] abind_1.4-5 compiler_4.3.2
[41] bit64_4.0.5 withr_2.5.2
[43] doParallel_1.0.17 ConsensusClusterPlus_1.66.0
[45] backports_1.4.1 BiocParallel_1.36.0
[47] viridis_0.6.4 DBI_1.2.0
[49] psych_2.3.12 ggstats_0.5.1
[51] dendextend_1.17.1 MASS_7.3-60
[53] DelayedArray_0.28.0 rjson_0.2.21
[55] scatterplot3d_0.3-44 flashClust_1.01-2
[57] tools_4.3.2 glue_1.6.2
[59] nlme_3.1-164 cluster_2.1.6
[61] generics_0.1.3 isoband_0.2.7
[63] gtable_0.3.4 tidyr_1.3.0
[65] hms_1.1.3 utf8_1.2.4
[67] XVector_0.42.0 foreach_1.5.2
[69] pillar_1.9.0 stringr_1.5.1
[71] emdbook_1.3.13 limma_3.58.1
[73] splines_4.3.2 logging_0.10-108
[75] circlize_0.4.15 dplyr_1.1.4
[77] lattice_0.22-5 bit_4.0.5
[79] tidyselect_1.2.0 ComplexHeatmap_2.18.0
[81] locfit_1.5-9.8 Biostrings_2.70.1
[83] knitr_1.45 gridExtra_2.3
[85] edgeR_4.0.2 xfun_0.41
[87] statmod_1.5.0 DT_0.31
[89] stringi_1.8.3 codetools_0.2-19
[91] bbmle_1.0.25.1 tibble_3.2.1
[93] multcompView_0.1-9 cli_3.6.2
[95] xtable_1.8-4 systemfonts_1.0.5
[97] munsell_0.5.0 Rcpp_1.0.11
[99] coda_0.19-4 png_0.1-8
[101] bdsmatrix_1.3-6 parallel_4.3.2
[103] leaps_3.1 blob_1.2.4
[105] prettyunits_1.2.0 bitops_1.0-7
[107] apeglm_1.24.0 viridisLite_0.4.2
[109] mvtnorm_1.2-4 scales_1.3.0
[111] purrr_1.0.2 crayon_1.5.2
[113] GetoptLong_1.0.5 rlang_1.1.2
[115] cowplot_1.1.2 KEGGREST_1.42.0
[117] mnormt_2.1.1
