Entering edit mode

Hello, I am running into an issue reading my c.tab files in ballgown from stringtie. Please see error message; Thanks for any help!
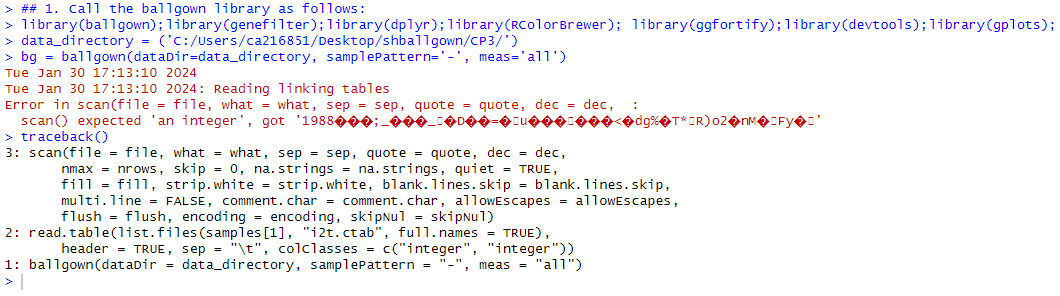
Hello, I am running into an issue reading my c.tab files in ballgown from stringtie. Please see error message; Thanks for any help!

The error is sort of self-explanatory - the expectation for i2t.ctab files is that they are two-column files with a header row and integer values, but your file does not contain integers. The error also seems to indicate that your files are binary instead of text (all those weird symbols are what you normally get if you try to parse a binary file as if it is text/numeric).
Why the i2t.ctab file(s) are that way is a different subject, and you will probably have to go back to how they were generated to figure that out.
Use of this site constitutes acceptance of our User Agreement and Privacy Policy.
Thank you for the reply, I appreciate any help. I tried on my personal computer and the error message is different now:
> bg = ballgown(dataDir=data_directory, samplePattern='-', meas='all') Wed Jan 31 19:10:24 2024 Wed Jan 31 19:10:24 2024: Reading linking tables Wed Jan 31 19:10:26 2024: Reading intron data files Wed Jan 31 19:10:32 2024: Merging intron data Wed Jan 31 19:10:33 2024: Reading exon data files Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec, : line 319196 did not have 12 elementsThat error indicates that you might have a truncated file. If you have access to Linux tools (on Windows you can install RTools to get them), you could do
To inspect the file in that region. But if it is as R says, then it's likely that something went wrong at that step, and the only thing you could do is re-run StringTie (or Cufflinks if you are really kicking it old school) to recapitulate the required file.
But anyway, the tuxedo suite has for the most part been superceded by more modern aligners like salmon and kallisto that perform pseudo-alignments to the transcriptome. It's much faster to use either of those tools, particularly if you are not trying to identify novel transcripts. You might consider switching if you aren't looking for novel transcripts.
Thank you James! I will try to align my data set with salmon, instead of the tuxedo method I was using. I appreciate your help.
Thank you, Candace