Entering edit mode

Hello,
I'm trying to use featureCounts to annotate a long read dataset obtained from nanopore sequencing. I noticed from the results reads that are supposed to be assigned unambiguously to a specific isoform, were actually assigned all different isoforms, clearly ignoring some isoform-specific splice junctions. One example is shown below, where all the reads were assigned to all 3 ACTG1 isoforms.
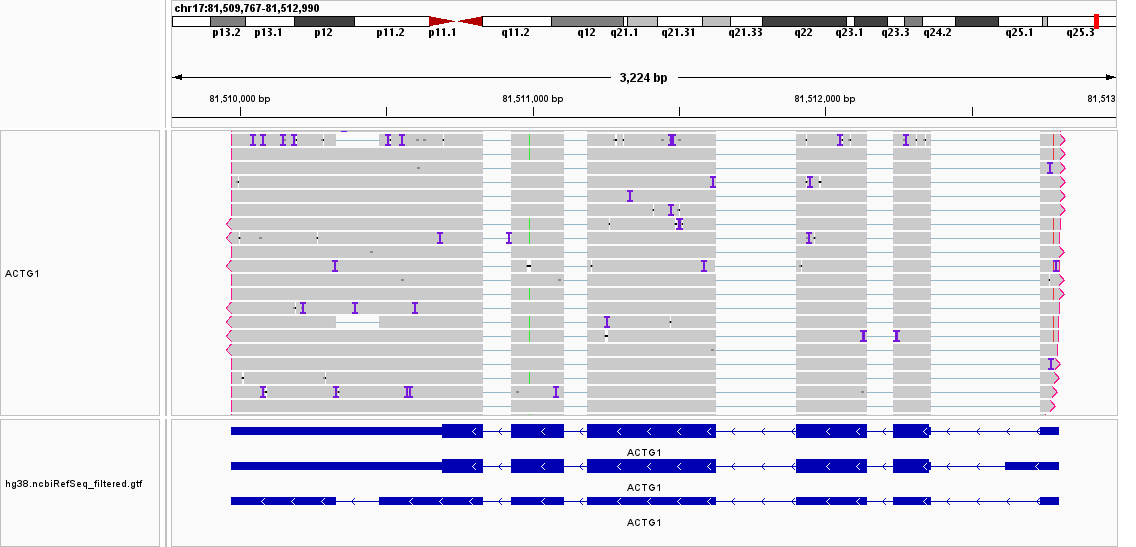
I was running featureCounts v2.0.6 in a Conda environment with the following command:
featureCounts -t exon -g transcript_id -O -R CORE -L -a ref.gtf -o counts.tab aln.bam
I was wondering what I can do about it, and I can send the dataset if needed.
Thank you!
