Entering edit mode

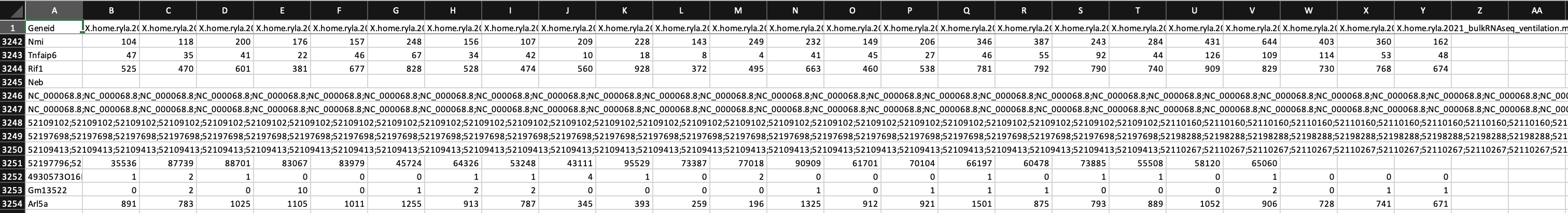
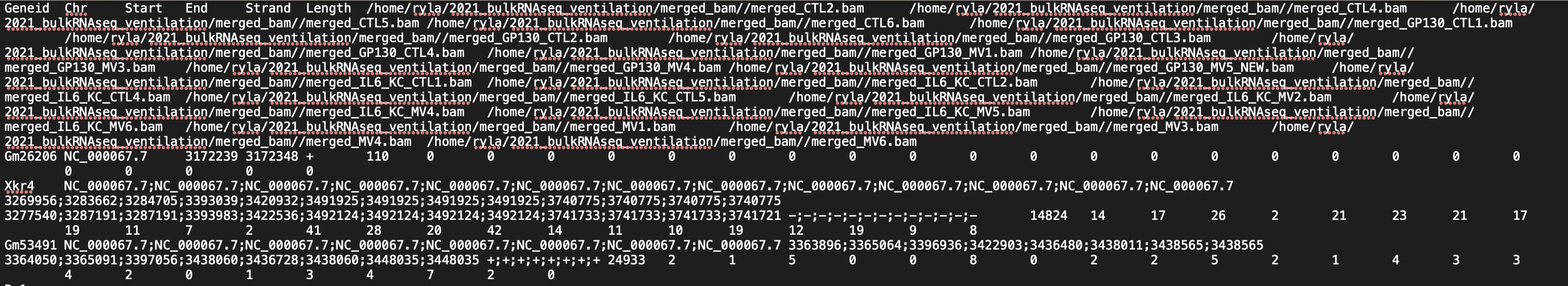
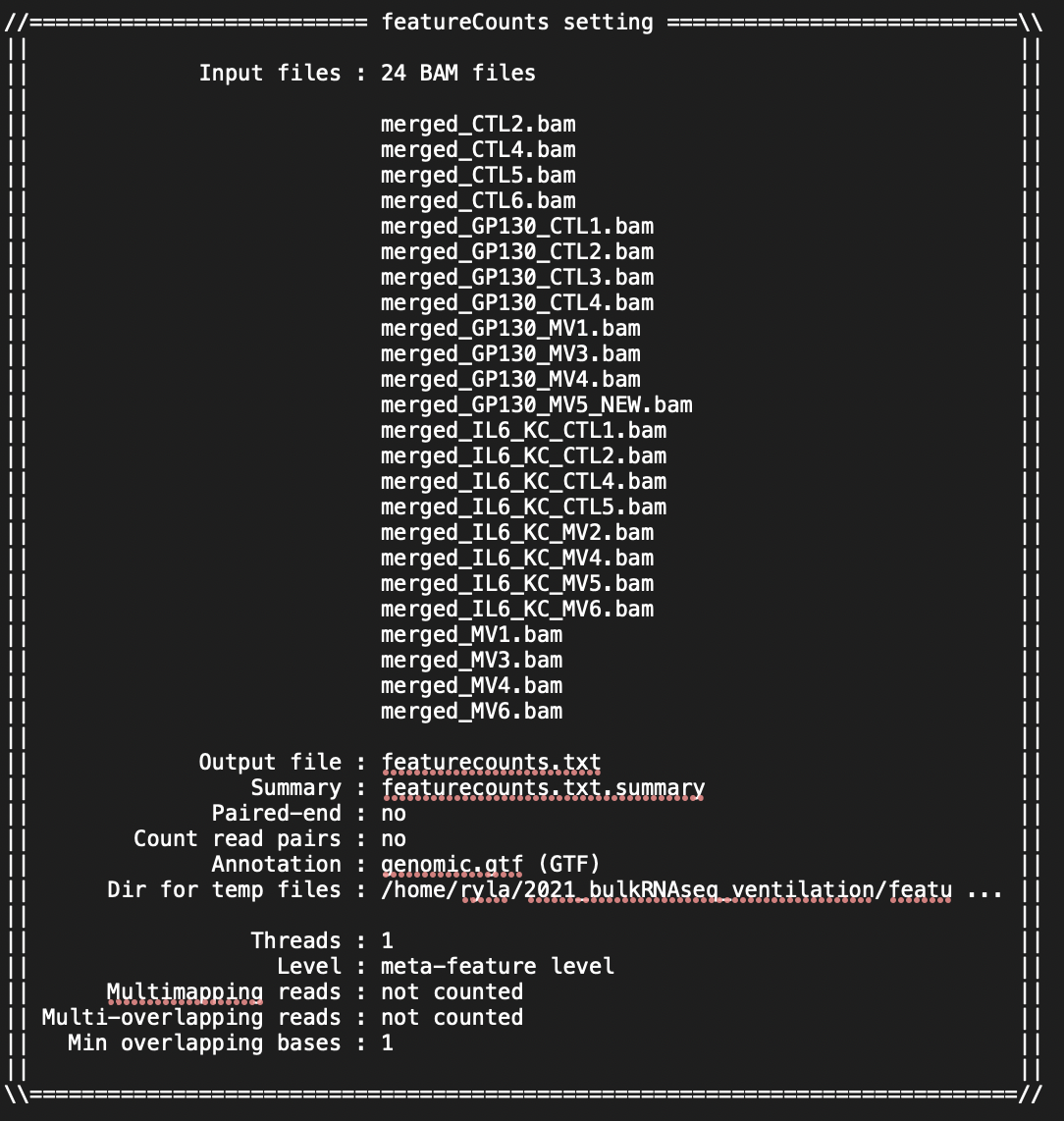
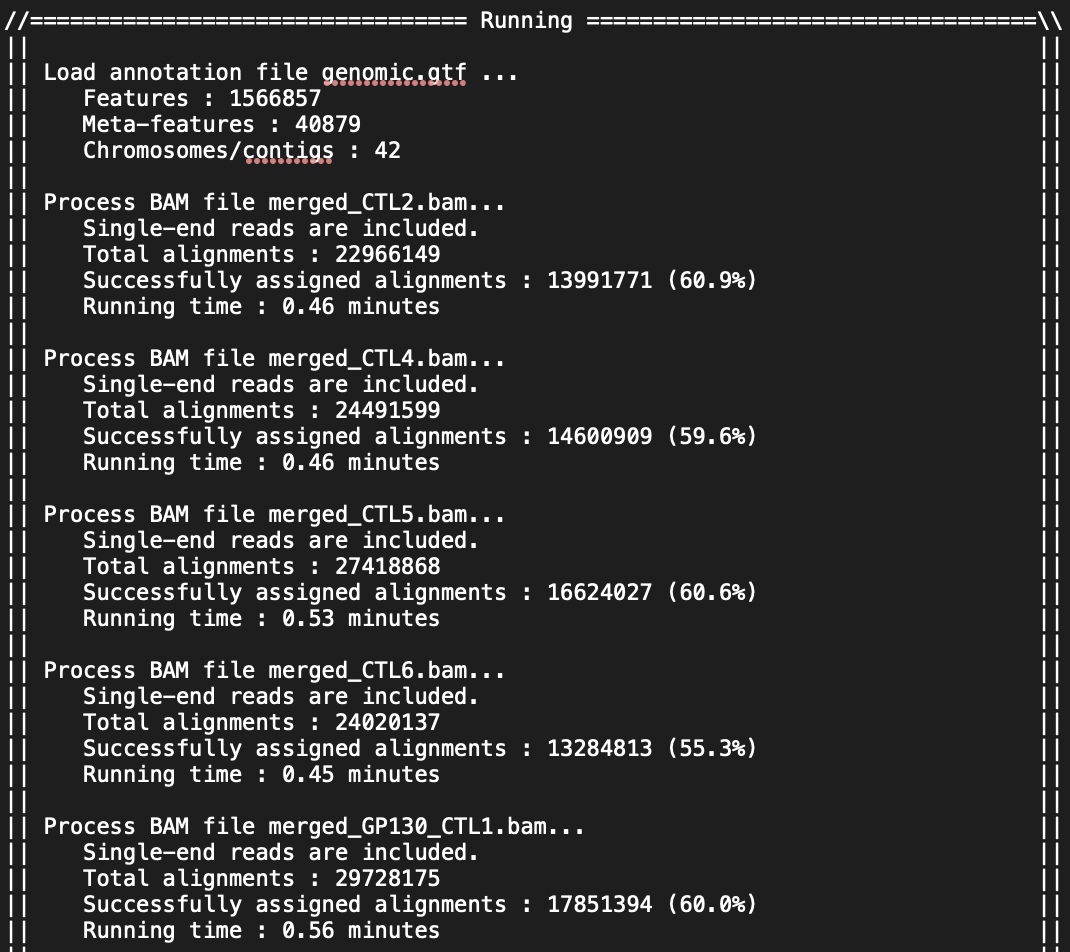
I am doing the analysis for RNASeq on 24 mouse samples (beginner to both coding and RNASeq analysis; I got the mouse GTF file from NCBI). I have attached screenshots of the summary of the featureCounts process as well as the .txt file I received as an output. Most of the columns of the .txt file look normal but there are a few that have blank columns from what looks to be a matrix alignment issue going from .txt to .xlsx in R. Am I missing something in this conversion or is there something wrong with my featureCounts output file? As you can see from the attachment, it did not get rid of the chromosome information and there are count values missing from a couple samples.
# Read text file, I have to specify fill because otherwise I get an error message: Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec, :
line 218 did not have 30 elements
data <- read.table("featurecounts.txt", header = TRUE, fill = TRUE)
# Omit columns 2 to 6
columns_to_keep <- c(1, (7:ncol(data)))
data_subset <- data[, columns_to_keep]
# Write as Excel file
write.xlsx(data_subset, "featurecounts_final.xlsx")