
Hi, Im peforming a miRNA-target prediction analysis, so I made use of the mirtarrnaseq library. Im a little naive at this kind of analysis and I dont know if im using the correct approach.
I have on one hand, genes from differential expression analysis (RNAseq) between patients and controls and, on the other hand, diferentialy expressed miRNAs between patients and controls (same samples). My idea was to find out if any of the differentialy expressed genes were targets of the differentialy expressed miRNAs. To this end, I used the 3rd way to conduct the analysis ("Part3 - Identify significant miRNA mRNA relationships for 2 time points" from the guide). Is it the correct way to do it? The obtained correlations allude to how much a target fits a particular miRNA?
Thanks, Maria
Hi,
So yes in this case you can use the third option and your approach is correct. Where you are using the fold change differences between the miRNAs and mRNAs and comparing that to a random background assumption of differences between all miRNAs and mRNA fold changes after sampling to see if specific miRNA-mRNA difference is significantly different enough or the difference is random. Your significant interactions demonstrate if there is enough statistical evidence to state your miRNA-mRNA difference ( up regulation of one and down regulation of the other) is not random.
Hope this was clear!
-Mercedeh
First of all, thank you so much for your reply.
So if I understood well, if I obtain a significant correlation between mRNA1 and a miRNA1, it means ,on the one hand, that mRNA1 is a target for miRNA1 and, on the other hand, that if miRNA1 is downregulated and mRNA1 is upregulated, that difference between fold changes is not random.
Thanks again, your library was really useful to me.
Maria
Yes that is correct
Hello, in line with the previous questions, when the miRNA-mRNA interaction analysis is performed (3 rd way to conduct the analysis), I obtain a dataframe with the correlation results. This dataframe consist of 8 columns. Regarding "value" column, what this value means?, the higher that value is the higher is the correlation between the mRNA and its miRNA?. And regarding "pvalue column", does it refers to the pearson correlation p-value between the mRNA and its miRNA?
Thanks, I hope I formulated the question properly,
Maria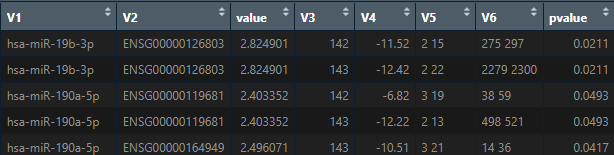
Hi Maria, so what is the code you are using? By the 3rd way to conduct the analysis do you mean looking at the interaction model from part 1 or do you mean you are looking at Part3- Identify significant miRNA mRNA relationships for 2 time points from fold changes? Please explain a bit more on which function you are running as each of these follows a different statistical test. Let me know and I am happy to help!
Hello, Im using "Part3- Identify significant miRNA mRNA relationships for 2 time points from fold changes". In mRNA data and miRNA data, there are two columns: One with the gene or miRNA name, respectively, and other with the fold change between same groups and same samples. I hope this information helps. Thanks for your help.
load data
File_mRNA <- file.choose() File_miRNA <- file.choose()
Expression data of mRNAS and miRNAS from condition v Control experiments.
mRNAs_data <- read.csv(File_mRNA, header = TRUE, sep = ";", dec = ",", row.names = 1) miRNAs_data <- read.csv(File_miRNA, header = TRUE, sep = ";", dec = ",", row.names = 1)
Estimate miRNA mRNA differences for your dataset.
inter0_D<- twoTimePoint(mRNAs_data, miRNAs_data)
make a background distribution for your miRNA mRNA FC diferences.
outs <- twoTimePointSamp(mRNAs_data,miRNAs_data, Shrounds = 10)
miRanda data import
miRanda <- getInputSpecies("Human1", threshold = 140)
Identify relationships below threshold.
signi_interR <- threshSigInter(inter0_D, outs)
miRanda intersection with results
results <- mirandaIntersectInter(signi_interR, outs, mRNAs_data, miRNAs_data, miRanda)
make data.frame results.
CorRes <- results$corrs
Hello Mercedeh, excuse me for not explaining it adecuately, Im using the "Part3 - Identify significant miRNA mRNA relationships for 2 time points".
The functions for performing this analysis are: getInputSpecies(), one2OneRnaMiRNA(), twoTimePoint(), twoTimePointSamp(), threshSigInter(), mirandaIntersectInter(), finInterResult(), drawInterPlots(), mirRnaHeatmapDiff().
The main functions would be:
What I wanted to know is what statistical test use this functions to stablish the significant relationships. Im sorry if it feels like a dumb question,
Kind regards, Maria
Maria please read the mirTarRnaSeq paper. In particular, Supplemental Table 2, clarifies all the statistical models, inputs and outputs. If you still have a question after that, let me know and happy to help.
https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08558-w#Sec23
Best,
-M