
Hello,
I am running into some perplexing behavior when using featureCounts. I would like to assign reads only to features with which they completely overlap (similar to HTSeq's intersection-strict set operation, and a functionality requested in a previous post). From reading the featureCounts documentation, I suspected that this would be accomplished by setting the nonOverlap parameter, described as "integer giving the maximum number of bases in a read (or a read pair) that are allowed not to overlap the assigned feature." to 0. For the rest of the post, I will be discussing the results when using the following call to featureCounts (where the goal was to assign reads to a gene with which it completely overlapped; the problems discussed here were encountered with 3 different calls to featureCounts with different feature assignments, namely assigning reads to genes, purely exonic regions, and purely exonic regions but assigning to the set of isoforms with which the read was consistent):
featureCounts -T 4 -s 1 -a Hs_genome_chr.gtf -R CORE -p --countReadPairs -g gene_id -t transcript --nonOverlap 0 --primary -o testout.featureCounts <input bam file>
While this worked seemingly as expected on several datasets, I ran into problem when assigning reads from a paired-end library with very small insert sizes and thus lots of instances of reads in a pair overlapping one another. In particular, the vast majority (> 90%) of read pairs from said library were not assigned, with featureCounts listing the reason for failed assignment being "Unassigned_Overlapping_Length". Increasing nonOverlap to 1 causes a small percentage of previously unassigned reads to be successfully assigned, and increasing it to 5 causes the majority of reads (~70%) to be successfully assigned. Below I have some IGV screenshots of examples, in all cases instances where the read completely and unambiguously overlaps an annotated gene that it should be assigned to:
Instances where read is assigned in all cases
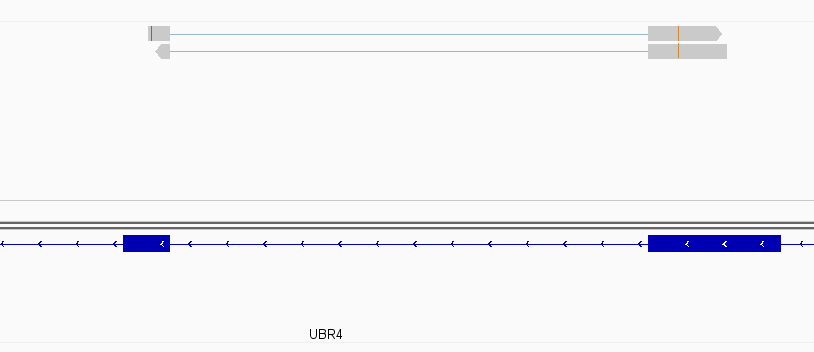
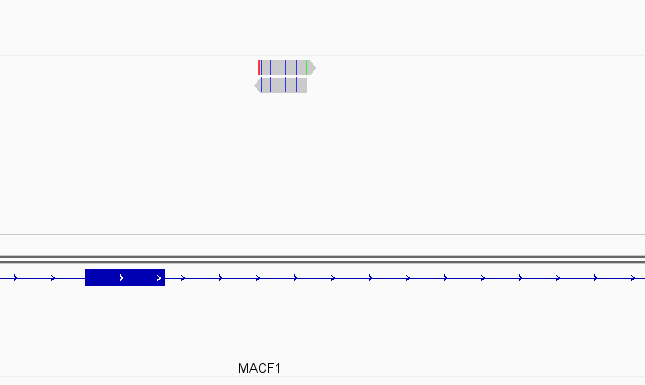
Instances where read is not assigned with nonOverlap = 0 but is assigned with nonOverlap = 1
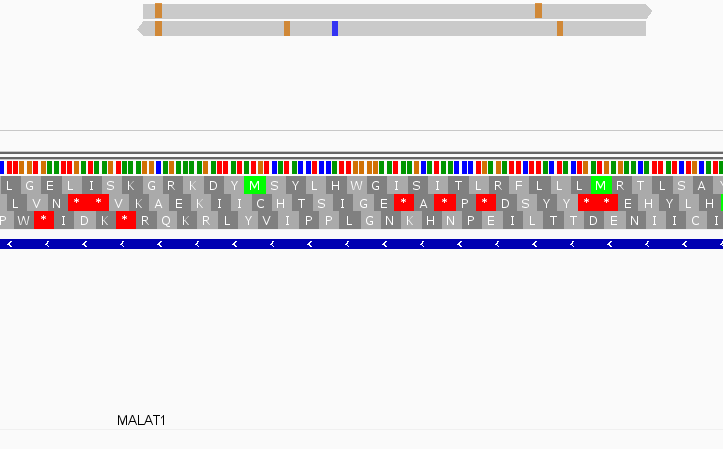
Instances where read is not assigned with nonOverlap = 0 and nonOverlap = 1, but is assigned with nonOverlap = 5
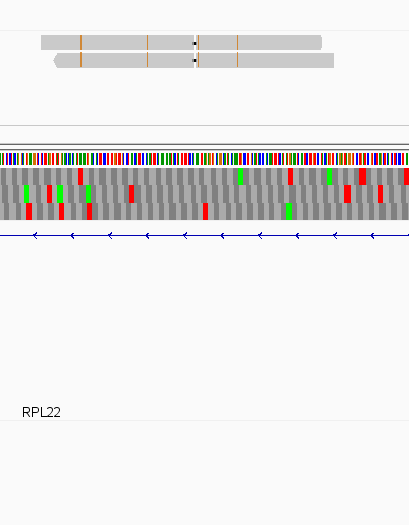
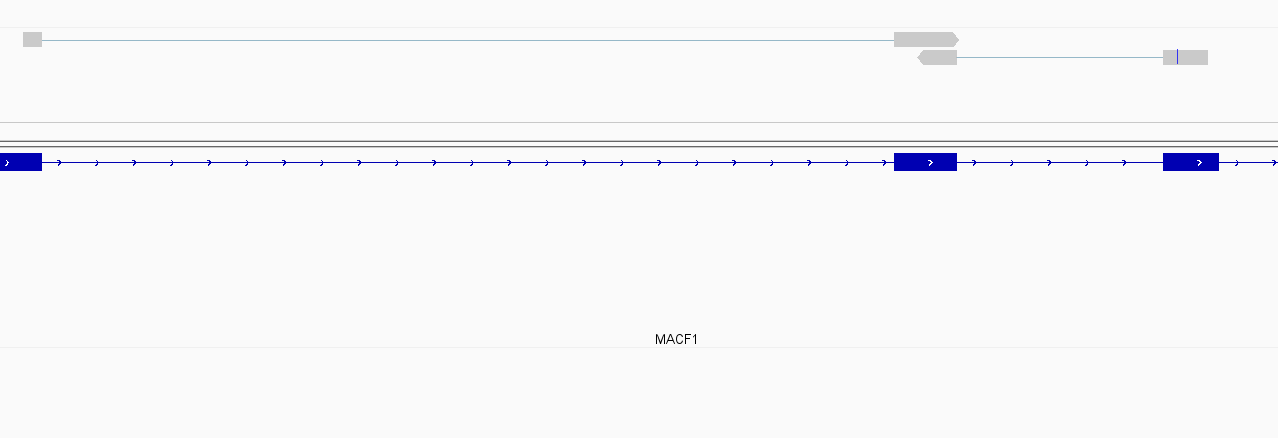
I have set up a Github repository with downsampled bam files containing reads that either 1) were assigned properly from the beginning, 2) were unassigned when nonOverlap = 0, 3) that were unassigned when nonOverlap=0 but assigned when nonOverlap=5 and 4) were unassigned when nonOverlap = 0 or nonOverlap = 1 but that were assigned when nonOverlap = 5.
The reads come from a stranded (FR) paired-end fastq file that was trimmed with fastp and aligned to the human genome with STAR. I am happy to provide any additional details.
Any explanation for why reads that clearly completely overlap a feature are not assigned when nonOverlap = 0 or 1 would be greatly appreciated!
Best, Isaac

This might be caused by your use of transcripts (
-t transcript) instead of exons for comparing read coordinates against feature coordinates. Can you try to use exons instead (this normally should be-t exonfor most GTF files) to see if you still have the same issue?You may also try to remove
-s 1and add-Oto rule out the possibility that this is caused by stranded counting or multi-overlapping (ie a read overlaps more than one gene).Thank you for your response. I can confirm that setting
-t exonand/or removing-s 1and adding-Ohas no impact on the number of successfully assigned reads.Best, Isaac