
Hello everyone and sorry in advance for the long post, I need some advices !
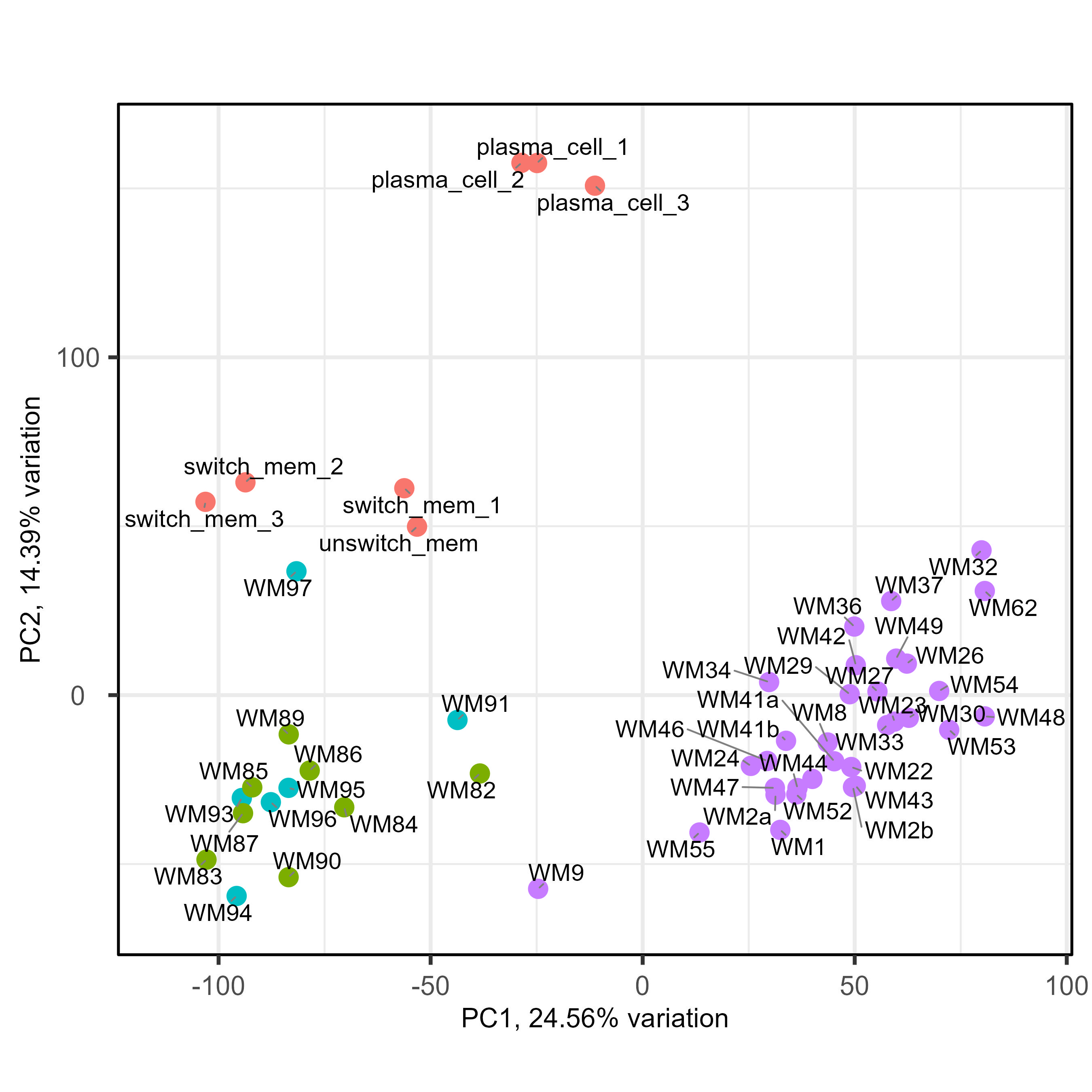
We have been fighting with a heavy batch & method effect in a cohort of bulk RNAseq from patients. As you can see below on my PCA (generated using all genes), I have three groups of samples : green and blue for patient samples done at the same place with one technology, purple for patient samples done at an other place with another technology, and red for two types of normal cells to compare patient with. As you can see the batch and method effect is very important... When doing a pairplots, I see that this batch effect can be seen the first and second PCs.
Sample_id Disease Mutation_SPI1 Mutation_MYD Secondary_Mutation Complete_mutation cohort cohort_for_RUVseq cohort_for_RUVseq_simpler
1 WM1 WM SPI1 MYD SPI1 MYD_SPI1 old_cohort old_cohort old_cohort
2 WM22 WM none MYD none MYD old_cohort old_cohort old_cohort
3 WM23 WM none MYD none MYD old_cohort old_cohort old_cohort
4 WM24 WM none MYD none MYD old_cohort old_cohort old_cohort
5 WM26 WM none MYD CXCR4 MYD_CXCR4 old_cohort old_cohort old_cohort
6 WM27 WM none MYD none MYD old_cohort old_cohort old_cohort
7 WM29 WM none MYD CXCR4_ARID1A MYD_CXCR4_ARID1A old_cohort old_cohort old_cohort
8 WM2a WM none MYD none MYD old_cohort old_cohort old_cohort
9 WM2b WM none MYD none MYD old_cohort old_cohort old_cohort
10 WM30 WM none MYD none none old_cohort old_cohort old_cohort
11 WM31 WM none MYD CXCR4 MYD_CXCR4 old_cohort old_cohort old_cohort
12 WM32 WM none MYD none MYD old_cohort old_cohort old_cohort
13 WM33 WM none MYD CXCR4 MYD_CXCR4 old_cohort old_cohort old_cohort
14 WM34 WM none MYD CXCR4 MYD_CXCR4 old_cohort old_cohort old_cohort
15 WM36 WM none MYD none MYD old_cohort old_cohort old_cohort
16 WM37 WM none MYD none MYD old_cohort old_cohort old_cohort
17 WM41a WM none MYD ARID1A MYD_ARID1A old_cohort old_cohort old_cohort
18 WM41b WM none MYD ARID1A MYD_ARID1A old_cohort old_cohort old_cohort
19 WM42 WM none MYD none MYD old_cohort old_cohort old_cohort
20 WM43 WM none MYD CXCR4 MYD_CXCR4 old_cohort old_cohort old_cohort
21 WM44 WM none MYD none none old_cohort old_cohort old_cohort
22 WM46 WM none MYD none none old_cohort old_cohort old_cohort
23 WM47 WM none MYD none MYD old_cohort old_cohort old_cohort
24 WM48 WM none none none none old_cohort old_cohort old_cohort
25 WM49 WM none none none MYD old_cohort old_cohort old_cohort
26 WM52 WM SPI1 MYD CXCR4_SPI1 MYD_CXCR4_SPI1 old_cohort old_cohort old_cohort
27 WM53 WM none MYD none MYD old_cohort old_cohort old_cohort
28 WM54 WM none MYD none none old_cohort old_cohort old_cohort
29 WM55 WM none MYD CXCR4 MYD_CXCR4 old_cohort old_cohort old_cohort
30 WM62 WM none MYD none MYD old_cohort old_cohort old_cohort
31 WM8 WM none MYD none MYD old_cohort old_cohort old_cohort
32 WM9 WM SPI1 MYD SPI1 MYD_SPI1 old_cohort old_cohort old_cohort
33 WM91 WM unknown unknown unknown unknown new_Novogene Novogene Novogene
34 WM93 WM unknown unknown unknown unknown new_Novogene Novogene Novogene
35 WM94 WM unknown unknown unknown unknown new_Novogene Novogene Novogene
36 WM95 WM unknown unknown unknown unknown new_Novogene Novogene Novogene
37 WM96 WM unknown unknown unknown unknown new_Novogene Novogene Novogene
38 WM97 WM unknown unknown unknown unknown new_Novogene Novogene Novogene
39 WM84 WM unknown unknown unknown unknown intermediate_Novogene Novogene Novogene
40 WM82 WM unknown unknown unknown unknown intermediate_Novogene Novogene Novogene
41 WM86 WM unknown unknown unknown unknown intermediate_Novogene Novogene Novogene
42 WM87 WM unknown unknown unknown unknown intermediate_Novogene Novogene Novogene
43 WM90 WM unknown unknown unknown unknown intermediate_Novogene Novogene Novogene
44 WM83 WM unknown unknown unknown unknown intermediate_Novogene Novogene Novogene
45 WM89 WM unknown unknown unknown unknown intermediate_Novogene Novogene Novogene
46 WM85 WM unknown unknown unknown unknown intermediate_Novogene Novogene Novogene
47 switch_mem_1 Healthy switch_mem switch_mem switch_mem switch_mem blueprint memory blueprint
48 switch_mem_2 Healthy switch_mem switch_mem switch_mem switch_mem blueprint memory blueprint
49 switch_mem_3 Healthy switch_mem switch_mem switch_mem switch_mem blueprint memory blueprint
50 unswitch_mem Healthy unswitch_mem unswitch_mem unswitch_mem unswitch_mem blueprint memory blueprint
51 plasma_cell_1 Healthy plasma_cell plasma_cell plasma_cell plasma_cell blueprint plasma_cell blueprint
52 plasma_cell_2 Healthy plasma_cell plasma_cell plasma_cell plasma_cell blueprint plasma_cell blueprint
53 plasma_cell_3 Healthy plasma_cell plasma_cell plasma_cell plasma_cell blueprint plasma_cell blueprint
So the idea we had was to use RUVseq, more precisely RUVg, to include correcting factor in the DESeq2 model, enabling us to correct the batch effect and compare freely samples between them. For RUVg, I tried different comparisons to determine a good dataset : purple vs only green and blue ; purple vs red vs green&blue ; purple vs green&blue vs red splitted in two (memory B cells and plasma cells). For the sake of clarity, I will only put below the code for the purple vs red vs green&blue. I tried different value of k for RUVg aswell. And then I know that I can include them like this in the DESeq2 model with "~ W1 + W2 + Complete_mutation". Here is my code :
dds <- DESeqDataSetFromTximport(txi.salmon,
design_file,
design = ~ cohort_for_RUVseq_simpler)
smallestGroupSize <- 3
keep <- rowSums(counts(dds) >= 10) >= smallestGroupSize
dds <- dds[keep,]
dds <- DESeq(dds)
vsd <- vst(dds, blind=FALSE)
res_oldcohort_novogene <- results(dds, contrast = c("cohort_for_RUVseq_simpler", "old_cohort", "Novogene"))
not_sig_oldcohort_novogene = rownames(res_oldcohort_novogene)[which(res_oldcohort_novogene$padj > 0.1)]
res_oldcohort_blueprint <- results(dds, contrast = c("cohort_for_RUVseq_simpler", "old_cohort", "blueprint"))
not_sig_oldcohort_blueprint = rownames(res_oldcohort_blueprint)[which(res_oldcohort_blueprint$padj > 0.1)]
res_Novogene_blueprint <- results(dds, contrast = c("cohort_for_RUVseq_simpler", "Novogene", "blueprint"))
not_sig_Novogene_blueprint = rownames(res_Novogene_blueprint)[which(res_Novogene_blueprint$padj > 0.1)]
not_sig_GLOBAL = Reduce(intersect, list(not_sig_oldcohort_novogene = not_sig_oldcohort_novogene,
not_sig_oldcohort_blueprint = not_sig_oldcohort_blueprint,
not_sig_Novogene_blueprint = not_sig_Novogene_blueprint)) # 2653 genes
## Run RUVg sequencing
table_to_see = as.data.frame(counts(dds))
RUVg_test <- newSeqExpressionSet(counts(dds))
RUVg_test_upper_quantile_norm <- betweenLaneNormalization(RUVg_test, which="upper")
empirical <- rownames(RUVg_test_upper_quantile_norm)[ rownames(RUVg_test_upper_quantile_norm) %in% not_sig_GLOBAL ]
correction_RUVg <- RUVg(RUVg_test_upper_quantile_norm, empirical, k=2)
pdat <- pData(correction_RUVg)
My problem is the read-out to say whether or not the correction and the factors extracted from RUVg worked properly. I have generated PCA plots colorizing the points of the PCA with the first two factors from RUVg. For the PCA (ntop = 500 and all genes), I have this for W1 from RUVg. It looks better with only the top 500 most variable genes (second picture) but this is not what I want I think...
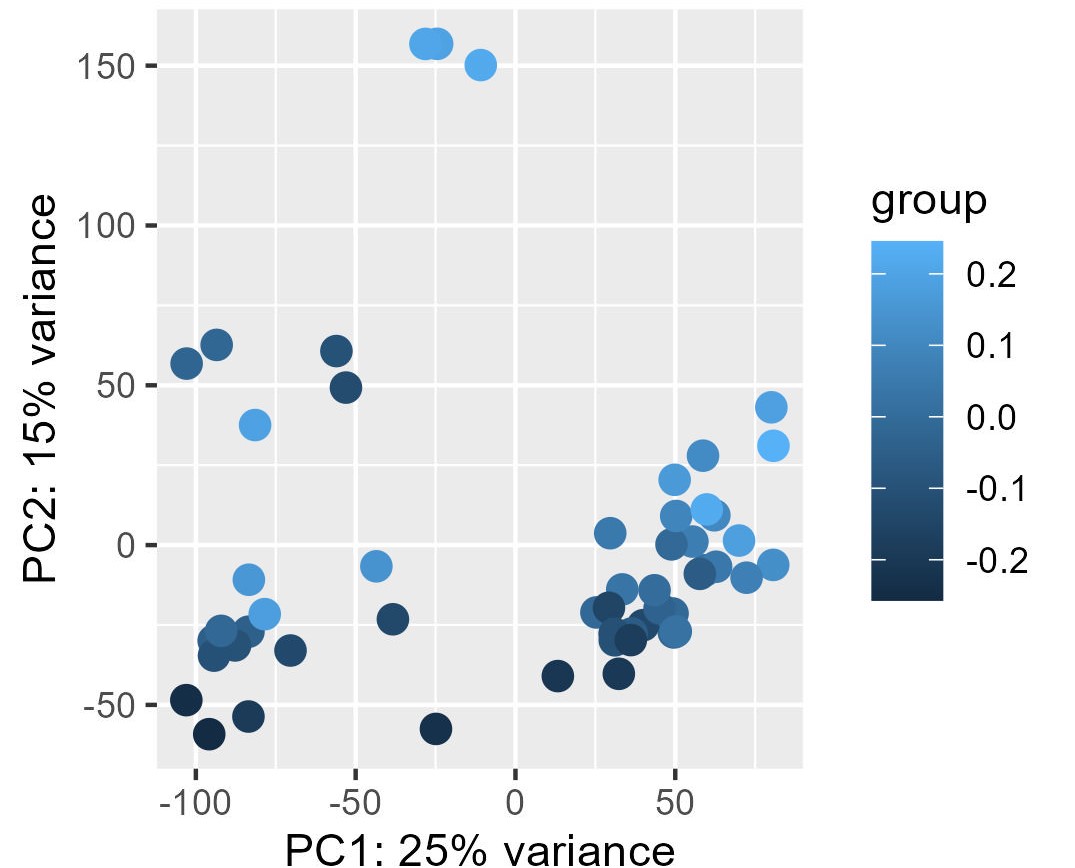
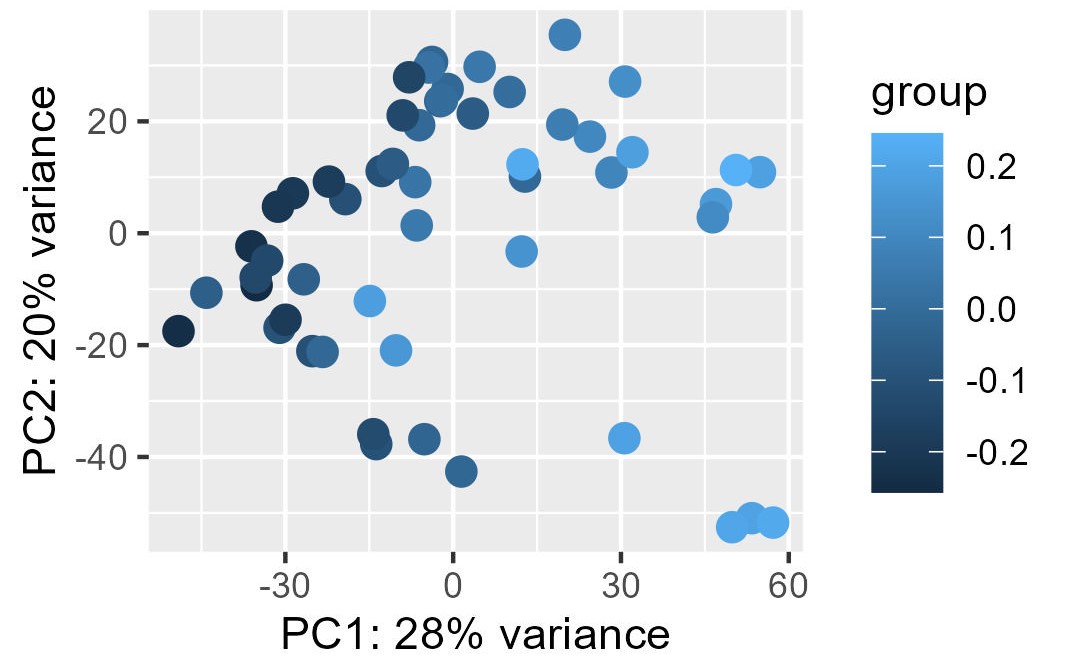
I also extracted for the different comparisons the correlation between the W1 and W2 RUVg factors and the coordinate on PC1 and PC2 (PCA with all genes). This can be find on this table.
correlations SIMPLER_comparison_simple
cor_W1_Dim1 0.172171
cor_W1_Dim2 0.6458019
cor_W2_Dim1 0.4765441
cor_W2_Dim2 -0.2840367
Finally, with this code here, I tried to remove this batch effect directly and see the PCA before (first picture) and after (second picture), which seems worst than without any correction.
empirical <- rownames(RUVg_test_upper_quantile_norm)[ rownames(RUVg_test_upper_quantile_norm) %in% not_sig_GLOBAL ]
correction_RUVg <- RUVg(RUVg_test_upper_quantile_norm, empirical, k=2)
pdat <- pData(correction_RUVg)
vsd <- vst(dds,blind=FALSE)
vsd_to_crush = vsd
mat <- assay(vsd)
mm <- model.matrix(~vsd$Complete_mutation, colData(vsd))
mat2 <- limma::removeBatchEffect(mat, covariates = pdat, design = mm)
assay(vsd_to_crush) = mat2
DESeq2::plotPCA(vsd, intgroup = "cohort", ntop = 100000)
DESeq2::plotPCA(vsd_to_crush, intgroup = "cohort", ntop = 100000)
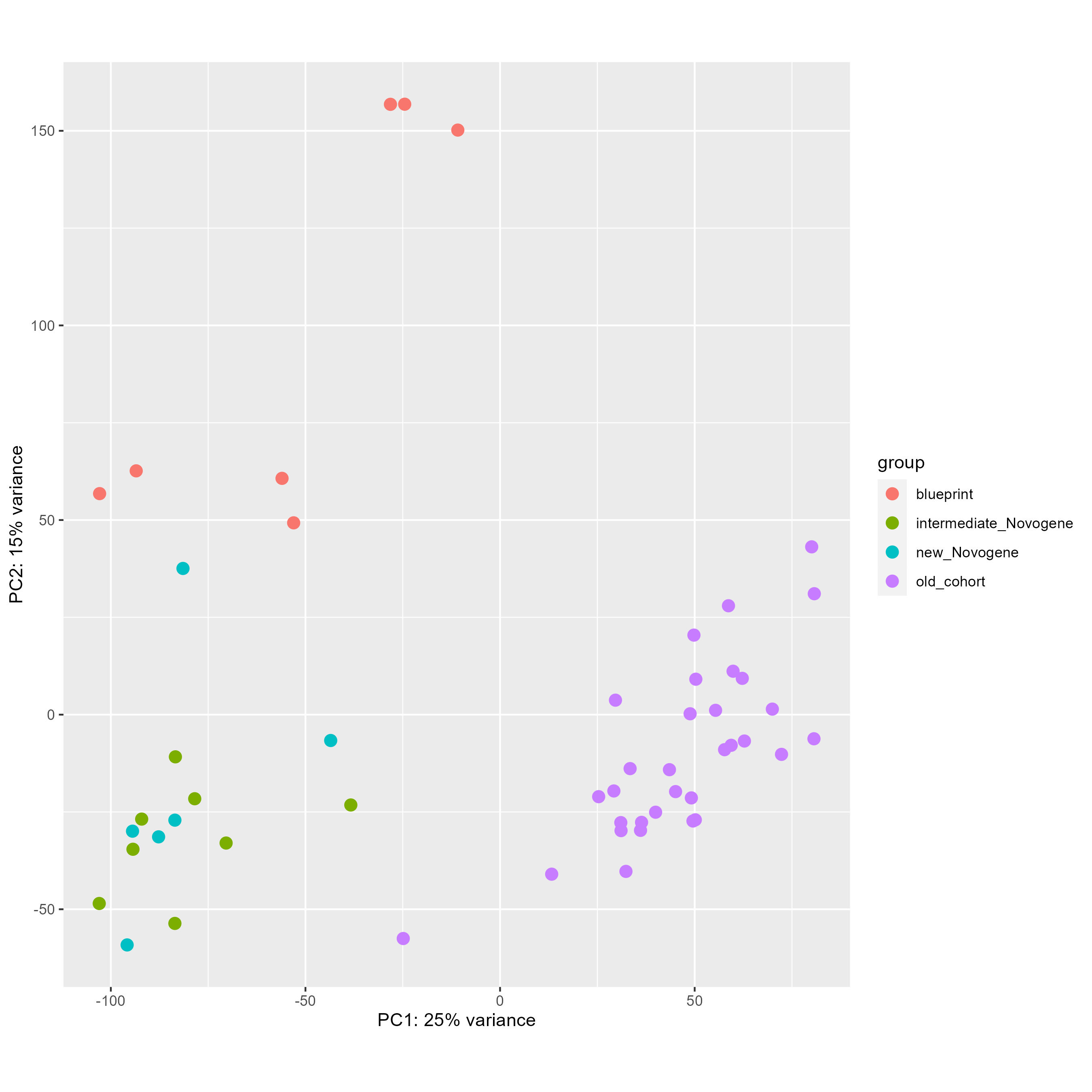
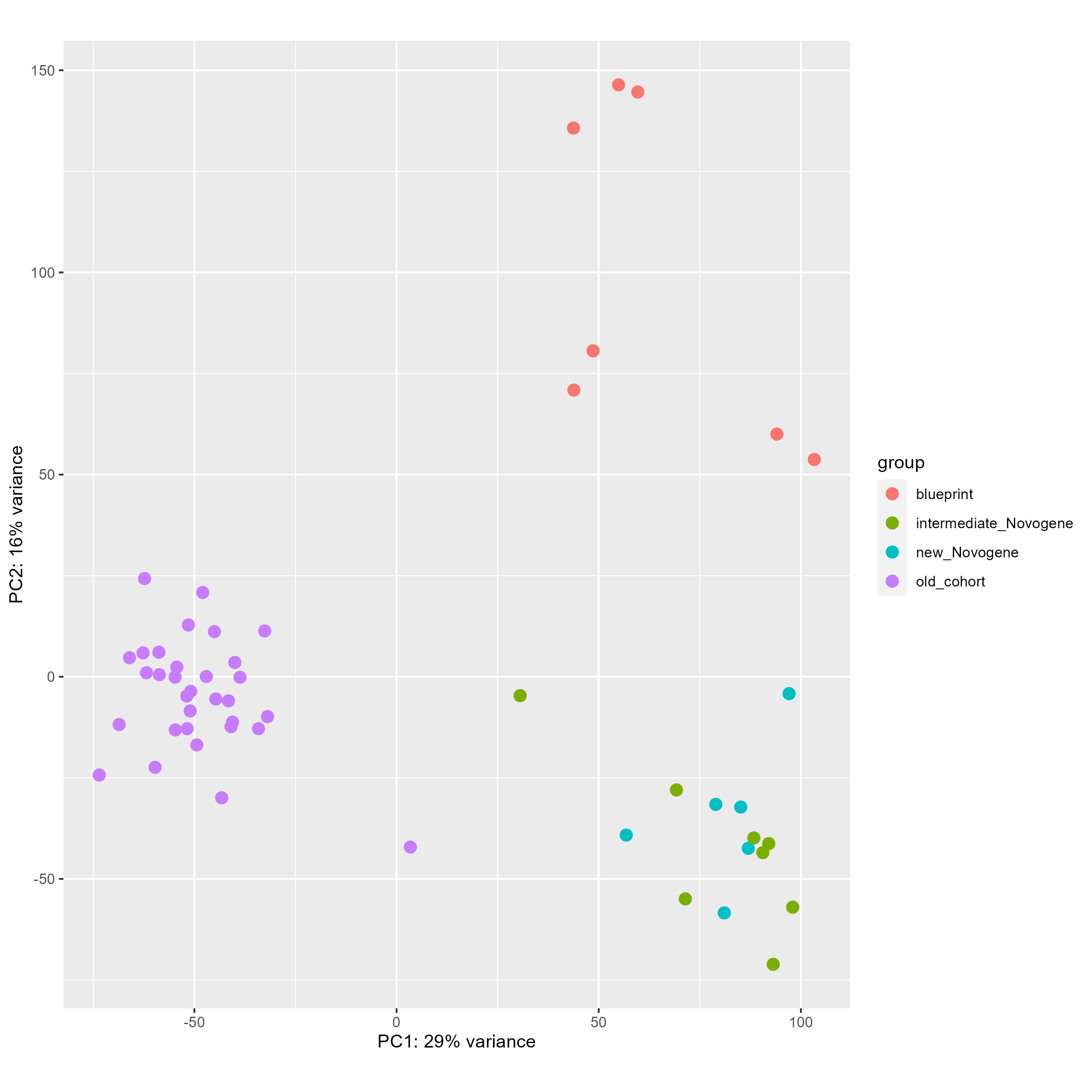
My questions are the following :
1) Was I right to dive directly into RUVseq and RUVg, and did I miss something that I could do that was simpler.
2) Which read-out for the RUVg method would you use ?
3) Did I make the limma removebatcheffect function work correctly or am I missing something ?
4) If I did what needed to be done to assess the RUVg method, what would you do ? Analyse the cohort separately ?
Thank you in advance for reading and for your precious help ! Alexandre

My code for the sessionInfo() . If the pictures are too big, I will downsize them. THank you again for reading my post !