
Hello,
I am trying to add ellipses to each experimental group in an RNASeq data using DESeq2. In this RNASeq experiment there are 4 genotypes and 2 treatments. I transformed my data using variance stabilizing transformation (vst) as shown in the code below. I group my data on the PCA plot using Genotype(shape) and Diet(color).
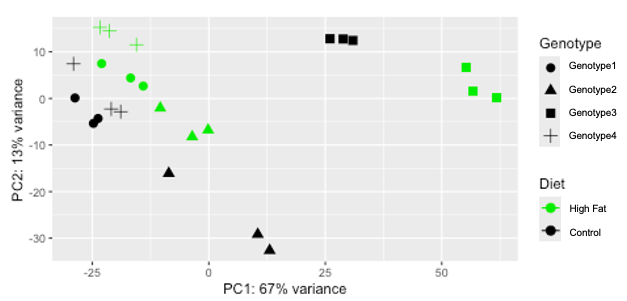
PCA plot I exported
# Constructing DESeqDataSet Object
rna_exp <- DESeqDataSetFromHTSeqCount(sampleTable = sampledesc, directory = "star", design = ~ Genotype_Diet)
# Fit DESeq2 statistical model
rna_exp2 <- DESeq(rna_exp)
# vst transformation
vsd <- vst(rna_exp2)
# calculate pcaData
pcaData <- plotPCA(vsd, intgroup= c("Diet", "Genotype"), returnData=TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
pcaData
# PC1 PC2 group Diet Genotype name
# Genotype1_Control_1 -18.9157077 -2.8444874 Control:Genotype1 Control Genotype1 Genotype1_Control_1
# Genotype1_Control_2 -20.9522176 -2.2563604 Control:Genotype1 Control Genotype1 Genotype1_Control_2
# Genotype1_Control_3 -29.0537285 7.4214088 Control:Genotype1 Control Genotype1 Genotype1_Control_3
# Genotype1_High_Fat_1 -21.3166182 14.4793876 High_Fat:Genotype1 High_Fat Genotype1 Genotype1_High_Fat_1
# Genotype1_High_Fat_2 -15.5327726 11.5241870 High_Fat:Genotype1 High_Fat Genotype1 Genotype1_High_Fat_2
# Genotype1_High_Fat_3 -23.3775377 15.2455884 High_Fat:Genotype1 High_Fat Genotype1 Genotype1_High_Fat_3
# Genotype2_Control_1 28.8351502 12.7642340 Control:Genotype1 Control Genotype1 Genotype2_Control_1
# Genotype2_Control_2 26.0118887 12.8089745 Control:Genotype1 Control Genotype1 Genotype2_Control_2
# Genotype2_Control_3 30.9264450 12.4476286 Control:Genotype1 Control Genotype1 Genotype2_Control_3
# Genotype2_High_Fat_1 55.2383247 6.6614633 High_Fat:Genotype1 High_Fat Genotype1 Genotype2_High_Fat_1
# Genotype2_High_Fat_2 61.7716660 0.1567968 High_Fat:Genotype1 High_Fat Genotype1 Genotype2_High_Fat_2
# Genotype2_High_Fat_3 56.7036605 1.5576229 High_Fat:Genotype1 High_Fat Genotype1 Genotype2_High_Fat_3
# Genotype3_Control_1 -23.7736534 -4.3032736 Control:Genotype3 Control Genotype3 Genotype3_Control_1
# Genotype3_Control_2 -28.7286469 0.1039441 Control:Genotype3 Control Genotype3 Genotype3_Control_2
# Genotype3_Control_3 -24.7687536 -5.3528158 Control:Genotype3 Control Genotype3 Genotype3_Control_3
# Genotype3_High_Fat_1 -23.0104137 7.4764965 High_Fat:Genotype3 High_Fat Genotype3 Genotype3_High_Fat_1
# Genotype3_High_Fat_2 -14.0798338 2.6576430 High_Fat:Genotype3 High_Fat Genotype3 Genotype3_High_Fat_2
# Genotype3_High_Fat_3 -16.7878296 4.3933782 High_Fat:Genotype3 High_Fat Genotype3 Genotype3_High_Fat_3
# Genotype4_Control_1 13.0572333 -32.6317159 Control:Genotype4 Control Genotype4 Genotype4_Control_1
# Genotype4_Control_2 10.4969905 -29.1774459 Control:Genotype4 Control Genotype4 Genotype4_Control_2
# Genotype4_Control_3 -8.5982418 -16.0843930 Control:Genotype4 Control Genotype4 Genotype4_Control_3
# Genotype4_High_Fat_1 -3.5673468 -8.2234753 High_Fat:Genotype4 High_Fat Genotype4 Genotype4_High_Fat_1
# Genotype4_High_Fat_2 -10.4241364 -2.0534517 High_Fat:Genotype4 High_Fat Genotype4 Genotype4_High_Fat_2
# Genotype4_High_Fat_3 -0.1539208 -6.7713347 High_Fat:Genotype4 High_Fat Genotype4 Genotype4_High_Fat_3
# PCA plot
png("PCA.png")
ggplot(pcaData, aes(PC1, PC2, color= Diet, shape= Genotype)) +
geom_point(size=3) +
xlab(paste0("PC1: ",percentVar[1],"% variance")) +
ylab(paste0("PC2: ",percentVar[2],"% variance")) +
coord_fixed() + scale_colour_manual(values = c(OP50 = "black", Glucose = "green2")) + stat_ellipse(type = "euclid")
dev.off()
Warning message:
I am unable to add ellipse because there are "too few points to calculate an ellipse".
Removed 8 rows containing missing values or values outside the scale range (geom_path()).
Question: Is there an alternative approach I should take to add ellipses to each genotype:Diet using the same color scheme I have?
I am interested in producing a pcaplot as shown below:
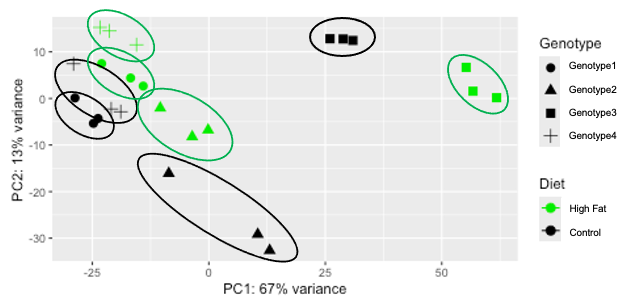
Thank you for any advice provided.
-robledoj13

Thank you for your the help @ATpoint.