It's probably not going to work that well using that species, but you can give it a shot. Note that you don't appear to have any good IDs. The closest is the column labeled EntrezID, which has LocusTags in it. You can convert those IDs using EDirect
esearch -db gene -query "Perca fluvialitis" | efetch -format docsum | xtract -pattern DocumentSummary -element Name Id OtherAliases OtherDesignations > tmp.txt
You can then use R
## just the first few - you will have to read them all in
> ids <- paste0("PFLUV_G", sprintf("%08d", c(184910,27280,227510,64610,172300,144370)))
> ids
[1] "PFLUV_G00184910" "PFLUV_G00027280" "PFLUV_G00227510" "PFLUV_G00064610"
[5] "PFLUV_G00172300" "PFLUV_G00144370"
## read in the file from EDirect and map
> mapper <- read.delim("tmp.txt", header = FALSE)
> head(mapper)
V1 V2 V3
1 saa 114559349 EPR50_G00004000
2 hsd11b2 120556064 PFLUV_G00038130
3 socs1a 114569781 SOCS-1, socs1
4 socs3a 114569548 EPR50_G00158030, SOCS-3, socs3
5 ahr2 114550691 EPR50_G00241230, AhR
6 tp53 114545465 EPR50_G00197220
V4
1 serum amyloid A-5 protein-like
2 LOW QUALITY PROTEIN: corticosteroid 11-beta-dehydrogenase isozyme 2|11-beta-hydroxysteroid dehydrogenase type 2
3 suppressor of cytokine signaling 1
4 suppressor of cytokine signaling 3
5 aryl hydrocarbon receptor-like
6 cellular tumor antigen p53-like|tumor suppressor p53
> egids <- mapper[match(ids, mapper[,3]),2]
> egids
[1] 120575335 120555898 120547723 120558550 120574289 NA
## now convert to Human using Orthology.eg.db
> library(Orthology.eg.db)
> select(Orthology.eg.db, egids, "Homo.sapiens","Perca.fluvialitis")
Perca.fluvialitis Homo.sapiens
1 120575335 NA
2 120555898 NA
3 120547723 NA
4 120558550 NA
5 120574289 NA
6 NA NA
It may simply be that the IDs I chose are not mapping (they are mostly LOC genes, meaning that they are probably a gene, but not identified), and you will get better results with the whole set, but it's probably going to be tough sledding.
There is an OrgDb for P. fluviatilis, but only a handful of the Gene IDs are mapped to any GO term, so that's a bust as well.

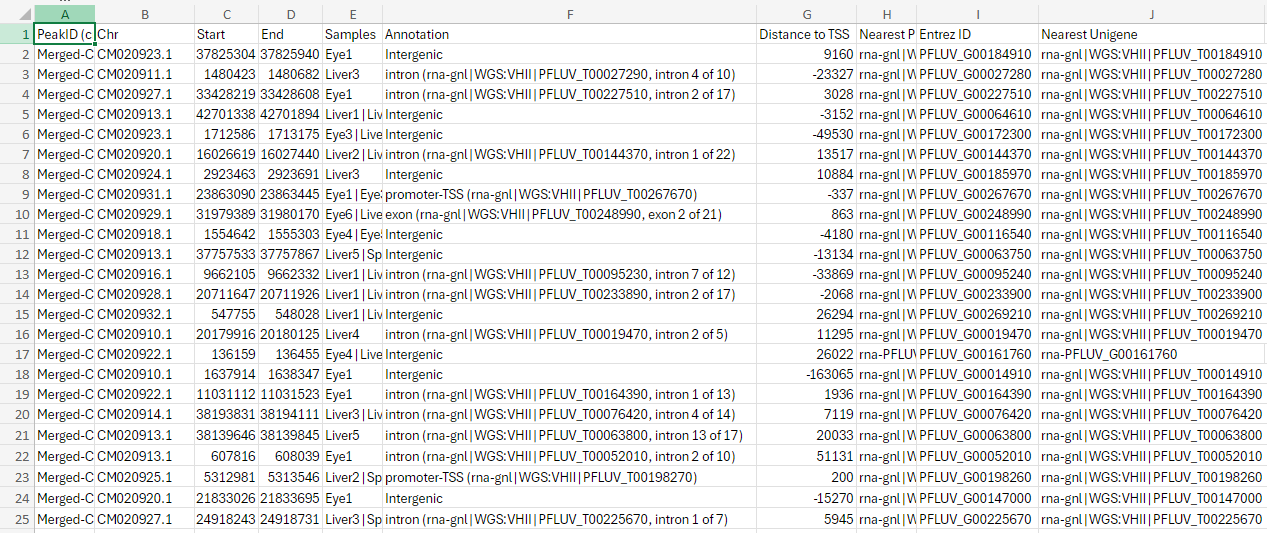

Not an answer to your question, but it seems the content of the column
Entrez IDare actually no entrez ids but rather aliases... Entrez ids consist of numbers only, but your ids start withPFLUV_.[Added: the ids starting with
PFLUV_are no aliases, but locus tags (see for example https://www.ncbi.nlm.nih.gov/gene/120575494); like James also noted below.]See: https://www.ncbi.nlm.nih.gov/gene/?term=txid8168[Organism:noexp]
You will then also notice that NCBI has info on ~32k genes of this species, and the function
makeOrgPackageFromNCBIfrom the packageAnnotationForgewill allow you to create a Bioconductor-compatibe annotation package for your species. See e.g. here for a recent thread on the use of this function: # Invalid keytype: GOALL. Please use the keytypes method to see a listing of valid arguments.There's already an
OrgDbfor this species on the AnnotationHub that hypothetically would be ideal, but there are almost no GO mappings, so it won't be very useful. Making anOrgDbmight work better though? I don't know how theOrgDbpackages are made for the AnnotationHub, but I sort of presume it's viamakeOrgPackageFromNCBI.