
Hello,
Both the GSVA module's implementation of ssGSEA and the Broad/GenePattern implementation (https://github.com/broadinstitute/ssGSEA2.0) are based on the papers from Barbie et al (1) and Abazeed et al (2), however, I cannot find a combination of parameters for which they produce identical results to each other.
For example, taking the random matrices a la the GSVA documentation:
set.seed(2023-11-09)
p <- 1000 ## number of genes
n <- 20 ## number of samples
ngs <- 10 ## number of gene sets
X <- matrix(rnorm(p*n), nrow=p,
dimnames=list(paste0("g", 1:p), paste0("s", 1:n)))
gs <- as.list(sample(10:100, size=ngs, replace=TRUE))
gs <- lapply(gs, function(n, p)
paste0("g", sample(1:p, size=n, replace=FALSE)), p)
names(gs) <- paste0("gs", 1:length(gs))
and running with the default parameters of GSVA's ssGSEA:
ssgseaPar <- GSVA::ssgseaParam(X, gs)
gsva_ssgsea <- GSVA::gsva(ssgseaPar, verbose=FALSE)
and running the broad implementation on the same data:
gct <- cmapR::GCT(mat=X)
gmt <- purrr::imap(gs, \(x, name) list(head=name, desc=name, len=length(x), entry=x))
cmapR::write_gct(gct, "X.gct", appenddim = FALSE)
cmapR::write_gmt(gmt, "gs.gmt")
ssGSEA2("X.gct", "compare/test01", "gs.gmt")
broad_ssgsea <- cmapR::parse_gctx("compare/test01-scores.gct")@mat
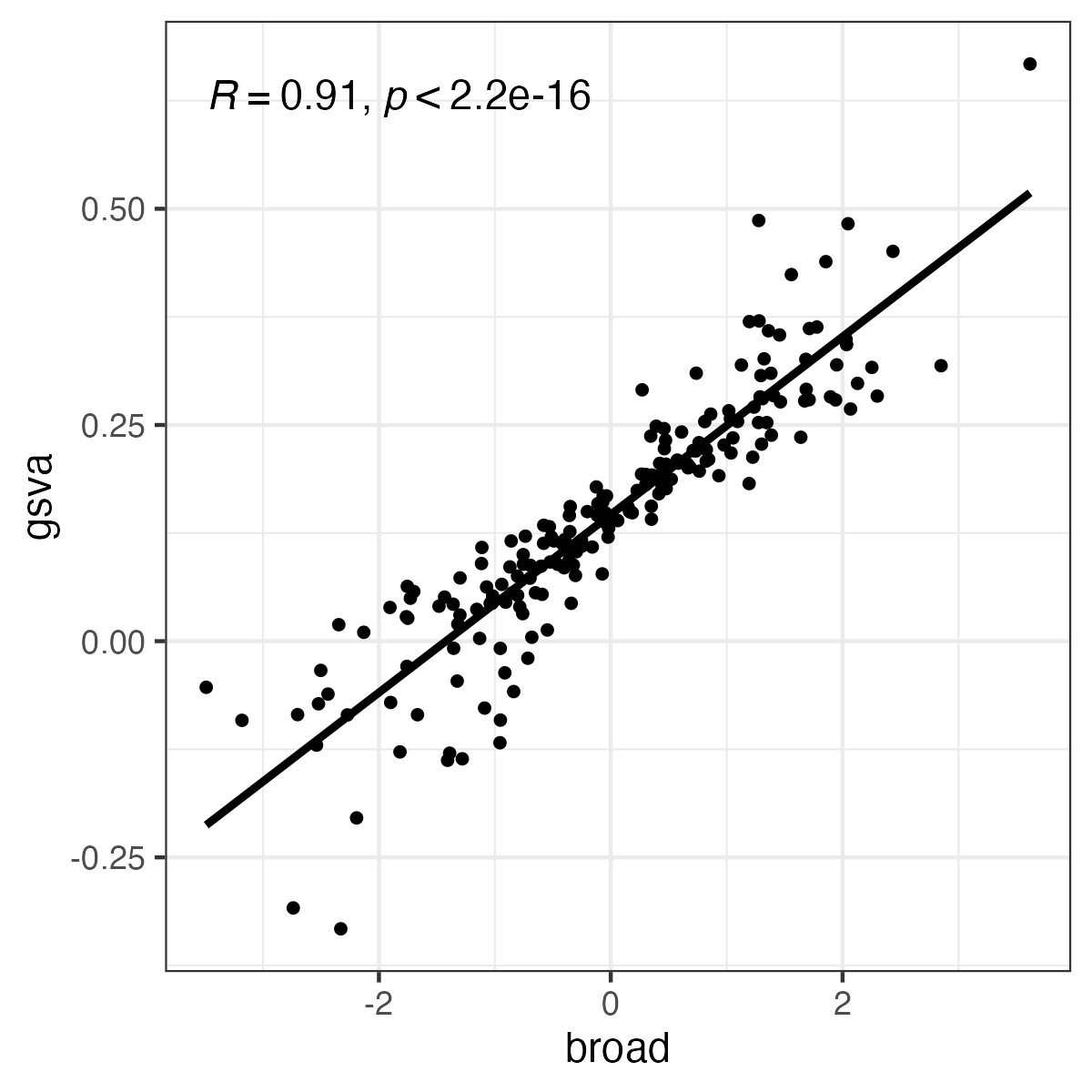
produces correlated but different results:
tibble::tibble(gsva=as.numeric(gsva_ssgsea), broad=as.numeric(broad_ssgsea)) |>
ggpubr::ggscatter("broad", "gsva", add = "reg.line", cor.coef = TRUE) +
ggplot2::theme_bw()
(See plot below). The critical parameters for GSVA seem to be normalize and alpha, but there seem to be no analogs in the Broad implementation.
Is there a way to make these two implementations behave identically?
Thank you!
sessionInfo( )
R version 4.4.0 (2024-04-24)
Platform: aarch64-apple-darwin23.2.0
Running under: macOS Sonoma 14.6.1
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Users/rzimmermann/.brew/Cellar/r/4.4.0_1/lib/R/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] magrittr_2.0.3 doParallel_1.0.17 iterators_1.0.14 foreach_1.5.2 verification_1.42 dtw_1.23-1 proxy_0.4-27 CircStats_0.2-6 MASS_7.3-61 boot_1.3-31 fields_16.3 viridisLite_0.4.2 spam_2.11-0 gtools_3.9.5 pacman_0.5.1
loaded via a namespace (and not attached):
[1] rstudioapi_0.16.0 jsonlite_1.8.9 magick_2.8.5 farver_2.1.2 ragg_1.3.3 fs_1.6.4 zlibbioc_1.50.0 vctrs_0.6.5 memoise_2.0.1 rstatix_0.7.2 htmltools_0.5.8.1 S4Arrays_1.4.1 usethis_3.0.0 broom_1.0.7 Rhdf5lib_1.26.0 Formula_1.2-5 SparseArray_1.4.8 rhdf5_2.48.0 htmlwidgets_1.6.4 cachem_1.1.0 mime_0.12 lifecycle_1.0.4 pkgconfig_2.0.3 rsvd_1.0.5 Matrix_1.7-0 R6_2.5.1 fastmap_1.2.0 GenomeInfoDbData_1.2.12 MatrixGenerics_1.16.0 shiny_1.9.1 digest_0.6.37 colorspace_2.1-1 AnnotationDbi_1.66.0 S4Vectors_0.42.1 rprojroot_2.0.4 irlba_2.3.5.1
[37] pkgload_1.4.0 textshaping_0.4.0 GenomicRanges_1.56.1 RSQLite_2.3.7 ggpubr_0.6.0 beachmat_2.20.0 labeling_0.4.3 cytolib_2.16.0 fansi_1.0.6 mgcv_1.9-1 httr_1.4.7 abind_1.4-8 compiler_4.4.0 remotes_2.5.0 bit64_4.5.2 withr_3.0.1 backports_1.5.0 BiocParallel_1.38.0 carData_3.0-5 DBI_1.2.3 pkgbuild_1.4.4 HDF5Array_1.32.1 maps_3.4.2 ggsignif_0.6.4 DelayedArray_0.30.1 sessioninfo_1.2.2 rjson_0.2.23 tools_4.4.0 httpuv_1.6.15 glue_1.8.0 nlme_3.1-166 rhdf5filters_1.16.0 promises_1.3.0 grid_4.4.0 cmapR_1.16.0 generics_0.1.3
[73] gtable_0.3.5 tzdb_0.4.0 tidyr_1.3.1 hms_1.1.3 car_3.1-3 BiocSingular_1.20.0 ScaledMatrix_1.12.0 utf8_1.2.4 XVector_0.44.0 BiocGenerics_0.50.0 pillar_1.9.0 stringr_1.5.1 vroom_1.6.5 GSVA_1.53.4 later_1.3.2 splines_4.4.0 flowCore_2.16.0 dplyr_1.1.4 lattice_0.22-6 bit_4.5.0 annotate_1.82.0 RProtoBufLib_2.16.0 tidyselect_1.2.1 SingleCellExperiment_1.26.0 Biostrings_2.72.1 miniUI_0.1.1.1 IRanges_2.38.1 SummarizedExperiment_1.34.0 stats4_4.4.0 Biobase_2.64.0 devtools_2.4.5 matrixStats_1.4.1 stringi_1.8.4 UCSC.utils_1.0.0 codetools_0.2-20 tibble_3.2.1
[109] BiocManager_1.30.25 graph_1.82.0 cli_3.6.3 systemfonts_1.1.0 xtable_1.8-4 munsell_0.5.1 Rcpp_1.0.13 GenomeInfoDb_1.40.1 png_0.1-8 XML_3.99-0.17 ellipsis_0.3.2 ggplot2_3.5.1 readr_2.1.5 blob_1.2.4 dotCall64_1.1-1 profvis_0.4.0 urlchecker_1.0.1 sparseMatrixStats_1.16.0 SpatialExperiment_1.14.0 GSEABase_1.66.0 scales_1.3.0 purrr_1.0.2 crayon_1.5.3 rlang_1.1.4 KEGGREST_1.44.1
Barbie, D. A., Tamayo, P., Boehm, J. S., Kim, S. Y., Susan, E., Dunn, I. F., . Hahn, W. C. (2010). Systematic RNA interference reveals that oncogenic KRAS- driven cancers require TBK1, Nature, 462(7269), 108-112. https://doi.org/10.1038/nature08460
Abazeed, M. E., Adams, D. J., Hurov, K. E., Tamayo, P., Creighton, C. J., Sonkin, D., et al. (2013). Integrative Radiogenomic Profiling of Squamous Cell Lung Cancer. Cancer Research, 73(20), 6289-6298. http://doi.org/10.1158/0008-5472.CAN-13-1616


Thanks for the prompt answer! I looked into the "original ssGSEA R-implementation" (https://github.com/GSEA-MSigDB/ssGSEA-gpmodule), unnormalized ssGSEA2 and unnormalized GSVA, and they seem to be near identical. The difference lies pretty much entirely in the normalization strategy. ssGSEA2 seems to use NES (which relies on permuting the gene sets), and GSVA ssGSEA uses the score range to normalize it. ssGSEA 1.0 does not have a post-projection normalization option. We do not need NES in particular.
I have one more question, though: I was wondering if Barbie et al 2009 proposed a rationale for their normalization. In the GSVA documentation, it says that this strategy is outlined on "(Barbie et al. 2009, online methods, pg. 2)". However I don't find this online methods in the current version (at least in front of the paywall). Do they justify it or just simply state that this is a way to normalize the scores?
Thanks again!
Thanks for your analysis of the differences between ssGSEA 2.0 and ssGSEA 1.0, as I said in my previous message, please file an issue in the GitHub repo if you would like to have version 2.0 as part of the GSVA package. Regarding how the normalization step is described in the original article, this is the sentence:
"Signature values were normalized using the entire set of 128 lung adenocarcinomas and 17 normal lung specimens."
located in the last page of the PDF version. If I recall correctly, because this happened more than 10 years ago, I looked up back then the Java code of the ssGSEA module in the GenePattern suite and found that normalization step done in the way GSVA is implementing it. You can email the authors of ssGSEA and I'm sure they'll be happy to provide you a PDF copy of their article.