Entering edit mode

I have been looking at dmpFinder function, and I was wondering if the function could work with beta values (as a matrix) and phenotypes (as a vector, for example race) without having the methylset or methylation object. My only worry is that how the function would be able to match the correct phenotypes to correct sample id beta values. Since the sample IDs are columns in the beta matrix, I am not sure what to do. Any suggestions would be appreciated. Thank you!

Hi James, Thank you a lot for responding. Hmm... so do you think if have matrix average_beta below: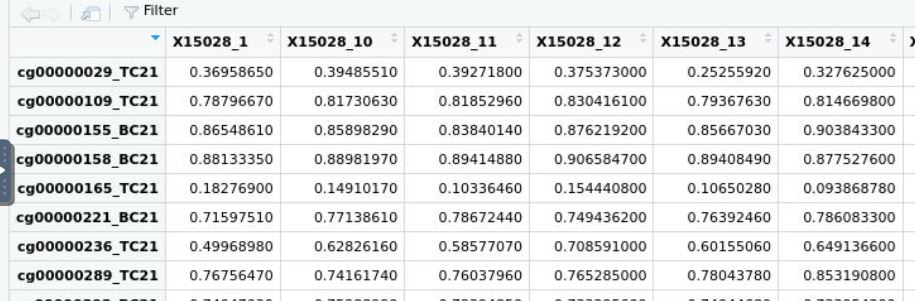
and the phenotype dataset below pheno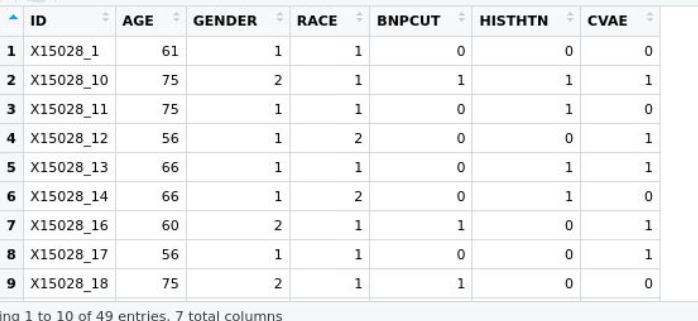
where both ids are matching in column and row, would dmpfiner give me a correct answer? Thank you! Neyousha
Yes, but you should probably use M-values instead of betas (convert using
logit2).However,
dmpFinderis just a way to allow people to uselmFitfromlimmato fit a model without having to figure out how to uselmFit. If you care to fit more than just one coefficient you will have to uselmFitdirectly.Thank you very much James, would you mind explaining why m-values instead of beta values? I have only beta_values available at this point.
You are better off using M-values instead of beta values because you are fitting a conventional linear regression, in which case it's better if the underlying distribution of your data is at least 'hump shaped'. This will be true for M-values, but not necessarily for beta values which are strictly between 0-1 and tend to be clustered at either extreme. If you have large enough N you can assume the central limit theorem will be in effect, in which case the underlying distribution doesn't matter (but the less hump-shaped, the larger N you need for the CLT to kick in).
It's just easier to defend using M-values because they are 'normal-ish' whereas beta values are not. If you really want to use betas, you might consider using
DSSwhich models the data assuming a beta distribution.But anyway, the
logit2function will convert your betas to M-values, so you can have M-values if you so desire/