
Hello,
Looking for peace of mind really. We routinely perform bulk ATAC across multiple primary human cell types (fibros, immune cells). We recently did bulk ATAC-seq on human mammary epithelial cells and performed differential analysis with DESeq2. The design is 2 biological replicates of a 5 time-point time series. The counting step was done with summarizeOverlaps against an IDR-cleaned list of MACS3-detected peaks. We then contrasted each time point relative to the reference at Day 0. I have never seen this before, but DESeq2 only found mostly peaks with increased counts at one time point (Day 7), with close to 0 peaks with reduced counts. I have never seen such an unbalanced distribution (see MA plot and heatmap attached) 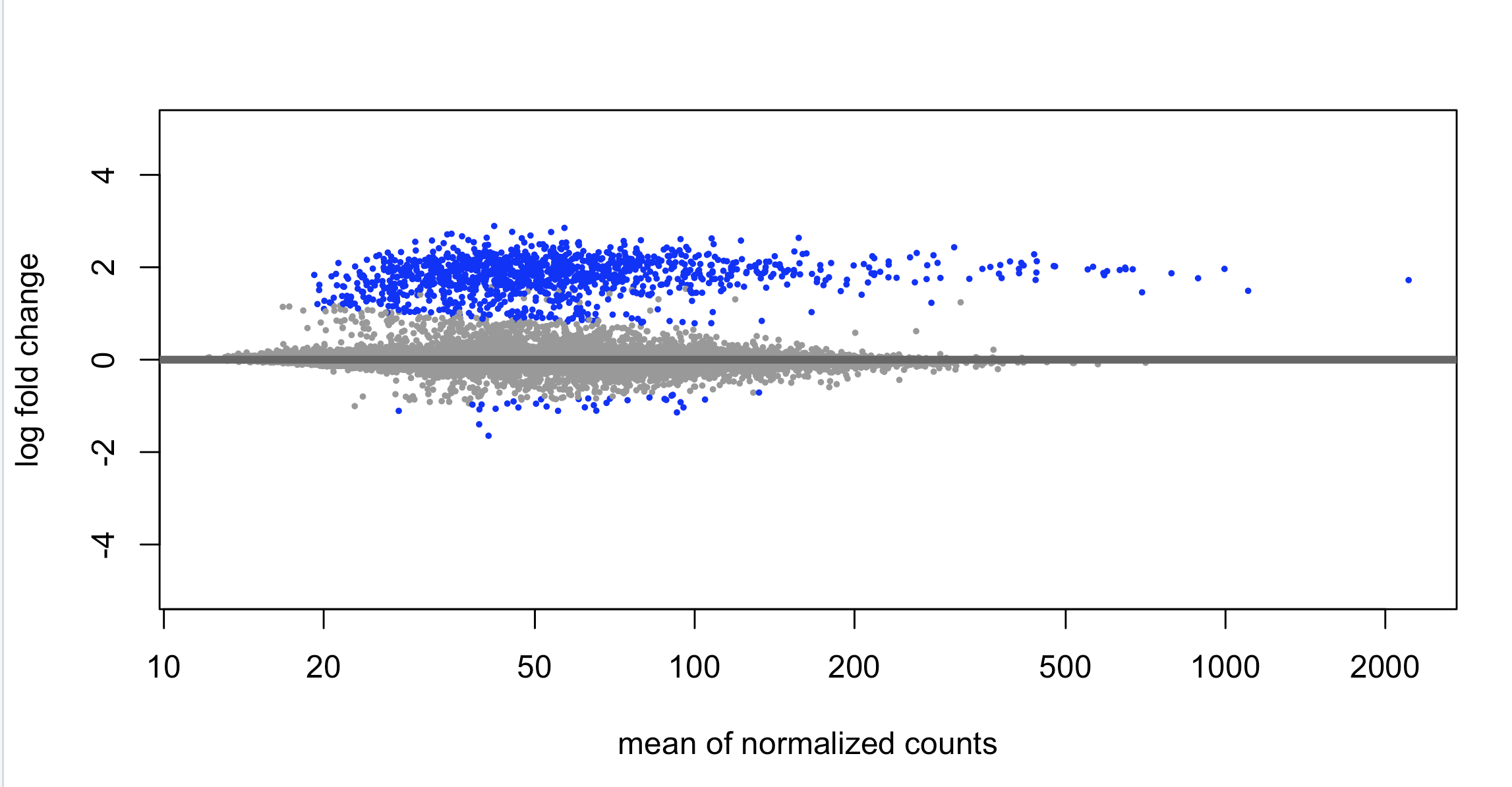
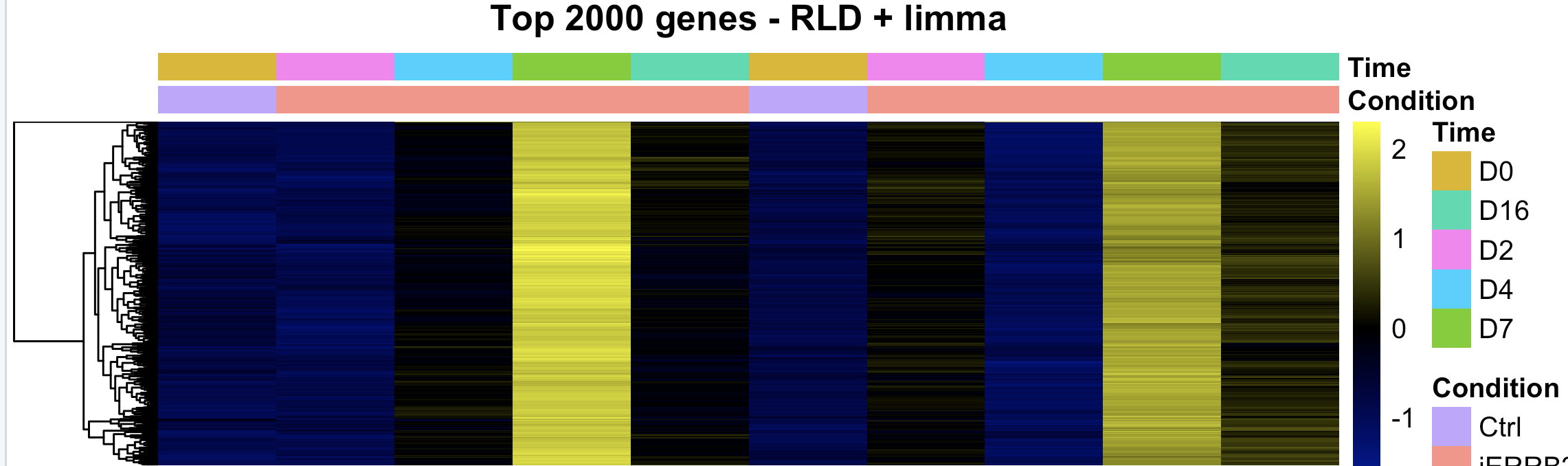
Cheers for any insight,
# include your problematic code here with any corresponding output
# please also include the results of running the following in an R session
sessionInfo( )

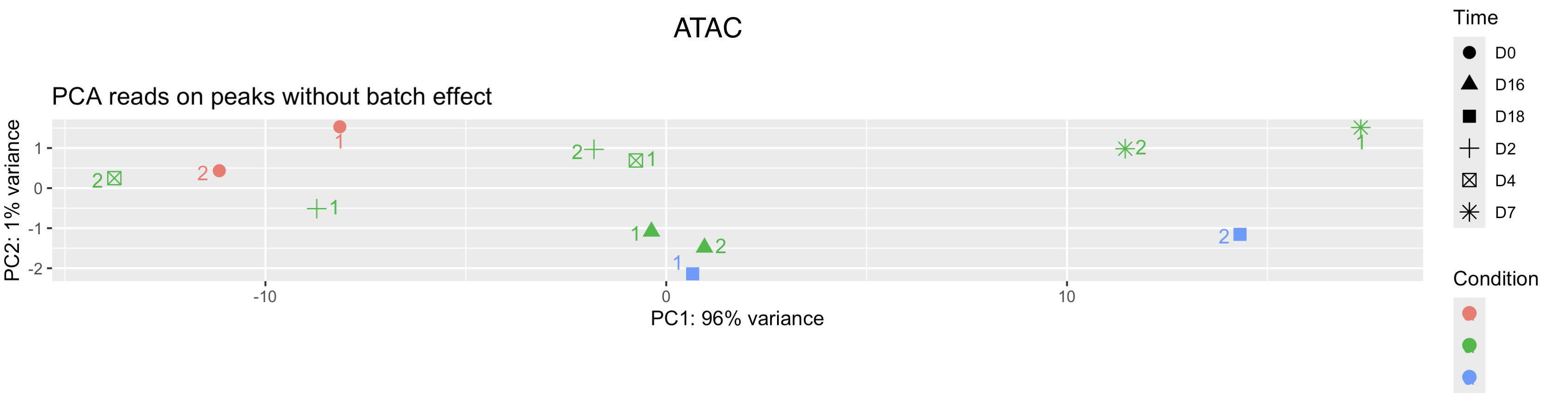 We will try adding two more replicates see if things get better.
We will try adding two more replicates see if things get better.
It is hard to comment here. My suggestion is:
results()