
Hello, I would like some assistance in figuring out why Rsubread and the bash subread are giving different feature count outputs. I came across this issue because I wrote a wrapper script for a pipeline used by a colleague, and during the validation process we have been trying to resolve why the wrapper feature count table differs from hers. The differences drastically affect the statistical analysis by DeSeq2. Through much testing, I found that our bam file outputs from STAR were exactly the same, and the issue arises when we run the files through featureCounts. The primary distinction is that her method uses Rsubread and the wrapper uses subread in bash. If I run her bam files through bash subread, our feature count tables are now identical, confirming that the divergence is occurring at the featureCounts step.
Neither of us was using the most updated versions of Rsubread and subread (Rsubread 2.18.0 and subread 2.0.1), and I recognized that this alone could cause an issue if those versions were not aligned with each other. So I downloaded Rsubread 2.20.0 and Subread 2.0.6 (unfortunately, I could not download 2.0.7 because it would require admin privileges, and 2.0.6 was the latest available on bioconda). I then ran the same 14 bam files (forward paired reads only) through Rsubread and bash Subread using the following scripts for each (same gtf file as well):
Subread 2.0.6 script:
featureCounts -t exon -g 'gene_id' -s 2 -o FeatureCounts_original.txt -a ${ref_dir}/${gtf_file} outputs/STAR/*/*.bam
Rsubread 2.20.0 script:
featureCounts(files=bamfiles, annot.ext=gtf, isGTFAnnotationFile=TRUE, isPairedEnd=FALSE, strandSpecific=2)
As far as I can tell by the manual, the parameters used between these two are the same, and both of the run logs indicate the same settings in the Settings box.
Of possible interest: the Rsubread output from version 2.18.0 was identical to that from 2.20.0, and the bash Subread output from version 2.0.1 was identical to that from 2.0.6.
In comparing the resulting feature count tables from the R version and bash version, there are major difference in the number of alignments assigned per sample and in total. Rsubread successfully assigned 205,861,649 more alignments, with an average of 14,704,404 more per sample. Here is a breakdown comparing the two outputs (information compiled from the run logs):
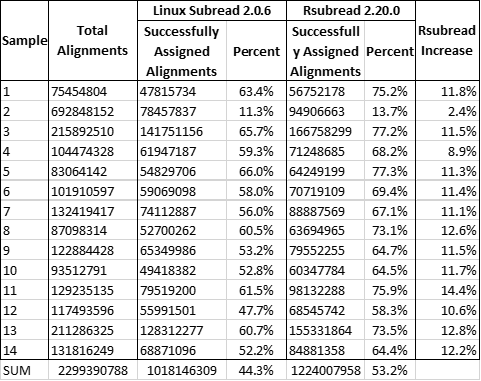
My team is very confused about which method is the truth, as we would draw vastly different conclusions about the populations of genes that are significantly differentially regulated from each method. Do you see anything that I am doing incorrectly or have suggestions for why the R and bash results are not in agreement? I am happy to provide more information as well. Thank you.
sessionInfo()
R version 4.4.0 (2024-04-24)
Platform: x86_64-pc-linux-gnu
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /apps/x86_64/R/4.4.0/lib64/R/lib/libRblas.so
LAPACK: /apps/x86_64/R/4.4.0/lib64/R/lib/libRlapack.so; LAPACK version 3.12.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: America/New_York
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices utils datasets methods base
loaded via a namespace (and not attached):
[1] compiler_4.4.0


Hi Gordon, Thank you so much for this response. I went back and checked, and you are absolutely correct that it was shown in the settings box, and I missed it. My eyes must have jumped oddly going back and forth between my terminals.
I greatly appreciate your commentary on the methods used as well, so that I can bring these to the attention of my team. I am currently a trainee, and my role in this has been to take my colleague's pipeline and wrap it to allow automation, but I kept the same parameters they previously used, so I am unsure of the reasoning behind the choices. However, I believe we are going to take a hard look at the best practices for this and make sure we are following them, and your advice will be very helpful.