If you get an error that says
Error in getCollapsedBetas(myAnnotation) :
could not find function "getCollapsedBetas"
It can only mean one of two things - either you have not loaded the package or the function is not exported from the package namespace. In this case it appears to be the former.
> library(DMRcate)
Setting options('download.file.method.GEOquery'='auto')
Setting options('GEOquery.inmemory.gpl'=FALSE)
> getCollapsedBetas
function (annot, ranges = NULL)
{
if (!is(annot, "CpGannotated")) {
stop("Error: annot is not a CpGannotated object. Please create one with cpg.annotate() or sequencing.annotate()")
}
if (!is.null(ranges)) {
if (!is(ranges, "GenomicRanges")) {
stop("Error: ranges is not a GRanges object")
}
}
betas <- annot@betas
if (!is.null(ranges)) {
idx <- annot@ranges %over% ranges
if (sum(idx) == 0) {
stop("Error: Ranges given do not overlap any CpGs in the given CpGannotated object.")
}
betas <- betas[idx, ]
}
betas
}
<bytecode: 0x00000232b482c898>
<environment: namespace:DMRcate>
As for the genes, consider the following, using example data.
> example(dmrcate)
> extractRanges(dmrcoutput)[203,]
snapshotDate(): 2024-10-24
see ?DMRcatedata and browseVignettes('DMRcatedata') for documentation
loading from cache
GRanges object with 1 range and 8 metadata columns:
seqnames ranges
<Rle> <IRanges>
[1] chr21 34813663-34813760
strand | no.cpgs
<Rle> | <integer>
[1] * | 3
min_smoothed_fdr Stouffer
<numeric> <numeric>
[1] 2.06921e-08 1.34603e-05
HMFDR Fisher
<numeric> <numeric>
[1] 8.83641e-09 3.83295e-08
maxdiff meandiff
<numeric> <numeric>
[1] 0 0
overlapping.genes
<character>
[1] IFNGR2, TMEM50B
## This DMR has two genes associated
> library(ggbio)
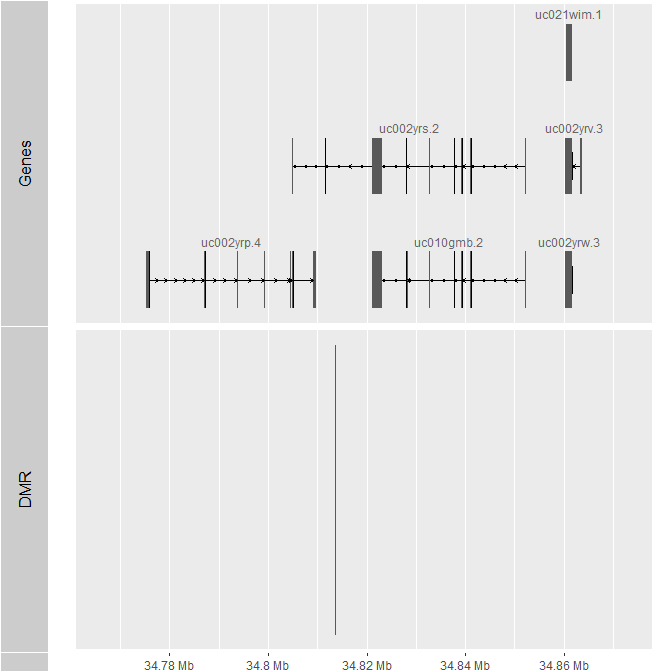
> tracks(list(Genes = autoplot(TxDb.Hsapiens.UCSC.hg19.knownGene, which = resize(GRanges("chr21:34813663-34813760"), 1e5, "center")), DMR = autoplot(extractRanges(dmrcoutput)[203,])))
Parsing transcripts...
Parsing exons...
Parsing cds...
Parsing utrs...
------exons...
------cdss...
------introns...
------utr...
aggregating...
Done
Constructing graphics...
Which generates this plot.
And if we do this
> library(Homo.sapiens)
> select(Homo.sapiens, c("uc002yrs.2","uc002yrp.4","uc010gmb.2"), "SYMBOL","TXNAME")
'select()' returned 1:1 mapping
between keys and columns
TXNAME SYMBOL
1 uc002yrs.2 TMEM50B
2 uc002yrp.4 IFNGR2
3 uc010gmb.2 TMEM50B
You can see that the DMR is upstream of both IFNGR2 and TMEM50B. Oddly enough though, this is based on hg19 (and the example data use EPICv2 data, which are on hg38), so I don't know if the CpG sites are being lifted over to hg19?


Thank you so much for the detailed explanation.
I have loaded the package and already used dmrcate and extractRanges functions. I had no problem. What should I do if the function is not exported?
And for my second question, I used hg19 annotation. Should I change it to hg38? As I am going to perform correlation analysis, I will use beta values of the genes from DMR analysis and fold changes (FC) from DEG analysis. Let's think about the DMR you have given. I have FC for TMEM50B and IFNGR2. For the DMR part, which gene should I take into consideration?
If
getCollapsedBetasisn't exported, you need to update. It's been exported since DMRcate_3.1.9 and the release version is 3.2.0You said you were using EPIC data, but didn't say what version. The original EPIC array is based on hg19, and the V2 version is hg38. I wouldn't change genomes in general, as liftOver is not really meant for lifting very short sequences. It's easier to just stick with the original target genome.
As for interpreting the data, that's your job as the analyst. The example I show above is quite complicated, as the DMR is upstream of two different genes and intronic in one of those genes as well. It's possible that the DMR is affecting one or both of those genes. And it's my understanding that intronic DMRs may have different effects on gene expression than upstream DMRs. But like I said, it's your analysis and up to you to interpret.
When I updated, the problem was solved. I used the getCollapsedBetas function.
My EPIC array data is the original one, so I'll stick with hg19.
I got the whole idea of what I should do. Thank you so much. I've been struggling a lot and there is no one in my work environment to help as no one has ever worked with DNA methylation or bioinformatics at all. I am glad that we have this support site and people like you who have been patiently answering our questions.