Unfortunately you did not show how you generated your input data (the object enrichment), nor did you show the output from sessionInfo().
To me it seems you are using an old version of R/Bioconductor.
Anyway, using the current version of Bioconductor with the example dataset:
> library(clusterProfiler)
> library(org.Hs.eg.db)
> library(enrichplot)
>
>
> ## example list of genes
> data(geneList, package="DOSE")
> genes <- names(geneList)[abs(geneList) > 2]
>
> res <- enrichKEGG(gene = genes,
+ organism = "hsa",
+ keyType = "kegg",
+ pvalueCutoff = 0.05,
+ pAdjustMethod = "BH",
+ minGSSize = 10,
+ maxGSSize = 500)
>
> ## to increase readability, convert entrezid into symbols
> ## also calculate pairwise similarities (is in essence only applicable
> ## to GO terms, but just to be sure
>
> res <- setReadable(res, OrgDb = org.Hs.eg.db, keyType="ENTREZID")
> res <- pairwise_termsim(res)
>
> ## cnetplot
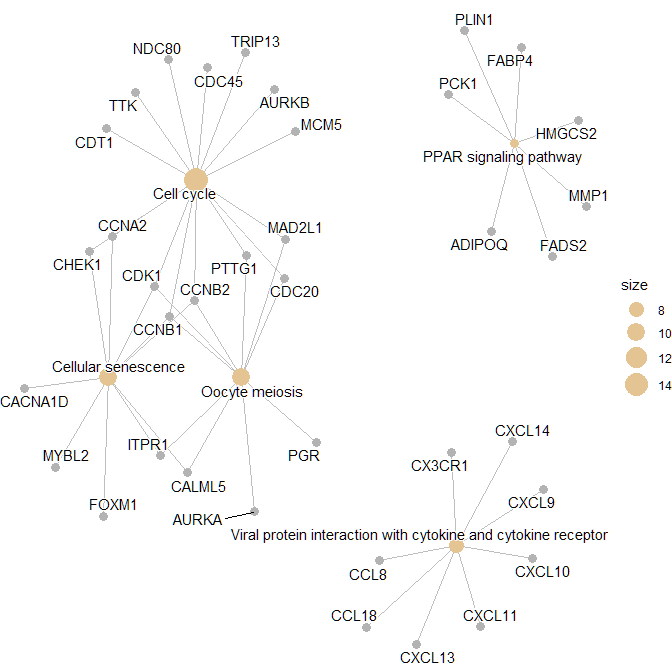
> p1 <- cnetplot(
+ res,
+ layout = igraph::layout_with_kk,
+ showCategory = 5,
+ color_category = "#E5C494",
+ size_category = 1,
+ color_item = "#B3B3B3",
+ size_item = 1,
+ color_edge = "grey",
+ size_edge = 0.5,
+ node_label = "all",
+ foldChange = NULL,
+ hilight_alpha = 0.3,
+ )
>
> print(p1)
>
> packageVersion("clusterProfiler")
[1] '4.14.6'
> packageVersion("enrichplot")
[1] '1.26.6'
>
>
>
> BiocManager::version()
[1] '3.20'
>
> sessionInfo()
R version 4.4.2 (2024-10-31 ucrt)
Platform: x86_64-w64-mingw32/x64
Running under: Windows 10 x64 (build 19042)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: Europe/Amsterdam
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] enrichplot_1.26.6 clusterProfiler_4.14.6 org.Hs.eg.db_3.20.0
[4] AnnotationDbi_1.68.0 IRanges_2.40.1 S4Vectors_0.44.0
[7] Biobase_2.66.0 BiocGenerics_0.52.0
loaded via a namespace (and not attached):
[1] tidyselect_1.2.1 dplyr_1.1.4 farver_2.1.2
[4] blob_1.2.4 R.utils_2.13.0 Biostrings_2.74.1
[7] lazyeval_0.2.2 fastmap_1.2.0 digest_0.6.37
[10] lifecycle_1.0.4 KEGGREST_1.46.0 tidytree_0.4.6
[13] RSQLite_2.3.9 magrittr_2.0.3 compiler_4.4.2
[16] rlang_1.1.5 tools_4.4.2 igraph_2.1.4
[19] data.table_1.17.0 ggtangle_0.0.6 labeling_0.4.3
[22] bit_4.6.0 gson_0.1.0 plyr_1.8.9
[25] RColorBrewer_1.1-3 aplot_0.2.5 BiocParallel_1.40.0
[28] withr_3.0.2 purrr_1.0.4 R.oo_1.27.0
[31] grid_4.4.2 GOSemSim_2.32.0 colorspace_2.1-1
[34] GO.db_3.20.0 ggplot2_3.5.1 scales_1.3.0
[37] cli_3.6.4 crayon_1.5.3 treeio_1.30.0
[40] generics_0.1.3 ggtree_3.14.0 httr_1.4.7
[43] reshape2_1.4.4 DBI_1.2.3 qvalue_2.38.0
[46] ape_5.8-1 cachem_1.1.0 DOSE_4.0.0
[49] stringr_1.5.1 zlibbioc_1.52.0 splines_4.4.2
[52] parallel_4.4.2 ggplotify_0.1.2 XVector_0.46.0
[55] yulab.utils_0.2.0 vctrs_0.6.5 Matrix_1.7-3
[58] jsonlite_1.9.1 gridGraphics_0.5-1 patchwork_1.3.0
[61] bit64_4.6.0-1 ggrepel_0.9.6 tidyr_1.3.1
[64] glue_1.8.0 codetools_0.2-20 cowplot_1.1.3
[67] stringi_1.8.4 gtable_0.3.6 GenomeInfoDb_1.42.3
[70] UCSC.utils_1.2.0 munsell_0.5.1 tibble_3.2.1
[73] pillar_1.10.1 fgsea_1.32.4 GenomeInfoDbData_1.2.13
[76] R6_2.6.1 lattice_0.22-6 R.methodsS3_1.8.2
[79] png_0.1-8 memoise_2.0.1 ggfun_0.1.8
[82] Rcpp_1.0.14 fastmatch_1.1-6 nlme_3.1-167
[85] fs_1.6.5 pkgconfig_2.0.3
>

